
Sarcomeric hypertrophic cardiomyopathy has heterogeneous phenotypic expressions, of which sudden cardiac death is the most feared. A genetic diagnosis is essential to identify subjects at risk in each family. The spectrum of disease-causing mutations in the Portuguese population is unknown.
MethodsSeventy-seven unrelated probands with hypertrophic cardiomyopathy were systematically screened for mutations by PCR and sequencing of five sarcomeric genes: MYBPC3, MYH7, TNNT2, TNNI3 and MYL2. Familial cosegregation analysis was performed in most patients.
ResultsThirty-four different mutations were identified in 41 (53%) index patients, 71% with familial hypertrophic cardiomyopathy. The most frequently involved gene was MYBPC3 (66%) with 22 different mutations (8 novel) in 27 patients, followed by MYH7 (22%), TNNT2 (12%) and TNNI3 (2.6%). In three patients (7%), two mutations were found in MYBPC3 and/or MYH7. Additionally, 276 relatives were screened, leading to the identification of a mean of three other affected relatives for each pedigree with the familial form of the disease.
ConclusionsDisease-associated mutations were identified mostly in familial hypertrophic cardiomyopathy, corroborating the idea that rarely studied genes may be implicated in sporadic forms. Private mutations are the rule, MYBPC3 being the most commonly involved gene. Mutations in MYBPC3 and MYH7 accounted for most cases of sarcomere-related disease. Multiple mutations in these genes may occur, which highlights the importance of screening both. The detection of novel mutations strongly suggests that all coding regions should be systematically screened. Genotyping in hypertrophic cardiomyopathy enables a more precise diagnosis of the disease, with implications for risk stratification and genetic counseling.
A miocardiopatia hipertrófica sarcomérica (MH) tem expressão fenotípica heterogénea, sendo a morte súbita cardíaca a mais temida. O diagnóstico genético é fundamental para identificar os indivíduos em risco em cada família. O perfil genético da doença é desconhecido na população portuguesa.
População e métodosEstudaram-se geneticamente 77 doentes-índice com MH, não-relacionados, pesquisando-se, por PCR e sequenciação, mutações nos genes MYBPC3, MYH7, TNNT2, TNNI3 e MYL2. Efetuou-se análise de co-segregação familiar na maioria dos doentes.
ResultadosIdentificaram-se 34 mutações diferentes em 41 doentes-índice (53%), 71% com MH familiar. O gene mais frequentemente envolvido foi o MYBPC3 (66%), identificando-se 22 mutações diferentes (8 novas) em 27 doentes-índice. Seguiram-se os genes MYH7 (22%), TNNT2 (12%) e TNNI3 (2,6%). Em 3 doentes (7%) identificaram-se 2 mutações nos genes MYBPC3 e/ou MYH7. Estudaram-se também 276 familiares detetando-se, em média, mais 3 indivíduos em risco, em cada pedigree com MH familiar.
ConclusõesForam identificadas mutações associadas a MH maioritariamente na doença familiar, reforçando a ideia corrente de as formas esporádicas estarem associadas a genes raramente estudados. A maioria das mutações é privada de cada família. O gene MYBPC3 é o mais frequentemente afetado. As mutações neste gene e no gene MYH7 são responsáveis pela maioria das situações de MH. Podem existir mutações múltiplas nestes genes, sendo mandatório o seu rastreio. A identificação de mutações novas aconselha a rastrear sistematicamente todas as regiões codificadoras. O estudo genético na MH permite um diagnóstico mais preciso da doença, determinante para a estratificação do risco e para o aconselhamento genético.
Sarcomeric hypertrophic cardiomyopathy (HCM) is the most prevalent genetic cardiovascular disease and one of the leading causes of sudden cardiac death, particularly in young people. The disease is defined by the presence of increased ventricular wall thickness or mass in the absence of loading conditions sufficient to cause the observed abnormality.1 The usual clinical marker of the disease is unexplained ventricular hypertrophy, usually identified on echocardiographic examination. Sarcomeric HCM is associated with mutations in genes encoding sarcomere or sarcomere-related proteins. Several different mutations associated with the disease have been identified in at least 12 genes, the myosin-binding protein C (MYBPC3) and beta-myosin heavy chain (MYH7) genes being the most common. Mutations in these two genes and also in the cardiac troponin T (TNNT2) and cardiac troponin I (TNNI3) genes are associated with the disease in up to 62% of unrelated sarcomeric HCM patients in various cohorts around the world.2–9
The disease has an autosomal dominant pattern of transmission, and its penetrance is variable and age-related. As hypertrophy may develop later in life, the commonly accepted prevalence of 0.2%10 may be an underestimation. The clinical course of HCM is highly unpredictable and there is a risk of sudden death, particularly in individuals considered to have a high clinical risk profile. The risk for those only genetically affected but with no phenotype (carriers) is not well defined.
Although it is believed that specific gene mutations with distinct functional characteristics may contribute to different phenotypes and prognosis, for most causal mutations prognostication will be difficult, given their private character in affected families. On the other hand, if it is assumed that the true prognostic value of HCM genetic testing simply relies on a positive test (identification of a pathogenic sarcomeric mutation)9 and not on the type or location of a particular mutation, then periodic evaluation for risk stratification is recommended to all genetically affected individuals.
Molecular diagnosis distinguishes between sarcomeric HCM and other hypertrophic heart disorders with distinct clinical courses and prognosis. The correct diagnosis of HCM should therefore be the first step in cases of unexplained left ventricular hypertrophy (LVH). If the genetic test is positive – a causal sarcomeric mutation is found in about 60% of affected individuals2 – a diagnosis is made and clinical workup is performed in order to help decision-making, particularly to prevent sudden death. Genetic screening of the family follows (in a cascade approach) to identify other at-risk family members. Absence of the causal mutation in relatives will reassure them, since in this case there is at present considered to be no risk of developing the disease.
In 30–40% of index patients, no mutation-associated disease is identified.11 In these cases, although the prognosis appears to be better,9 all first-degree relatives should be considered at risk and serial clinical screening is needed.
HCM is a global disease and genotype profiles regarding HCM-associated mutations have been described for several HCM populations in different countries worldwide.4–9,12–15 However, data on the Portuguese population are scarce16,17 and the prevalence of the most commonly involved genes is unknown. The main objective of this paper is to report the mutation profile in a Portuguese cohort of unrelated consecutive patients with sarcomeric HCM, followed at a large referral center.
MethodsPopulation and clinical dataThe study included 77 unrelated Portuguese index patients, most of Caucasian origin, 37 female, 40 male, aged 57 (22–82) years old at the time of genotyping, with a diagnosis of HCM based on major echocardiographic criteria as described elsewhere5,18 and defined by maximal end-diastolic left ventricular wall thickness (LVWT)>15mm. This population had a mean age at diagnosis of 44 (16–79) years and have been followed for a mean of 12.7 (1–35) years. Forty patients had at least 10 years of follow-up and 20 had more than 20 years. Most have been followed in the center; the others were referred for the first time to the center and a new echocardiogram and ECG were then performed.
A total of 314 relatives underwent a detailed clinical evaluation to determine whether HCM had a familial or sporadic character. Genotyping was performed in 276 of them in a cascade approach. A complete clinical assessment was undertaken in all patients, including a detailed family history with a pedigree including at least three generations. All available past exams (from index patients and relatives) were reviewed, including autopsy data on relatives who suffered sudden death. In addition to clinical assessment, a biochemical panel was performed in all index patients to exclude unrecognized muscle or renal involvement.
On the basis of clinical criteria and prior to genetic diagnosis, HCM was initially classified as familial, sporadic or inconclusive. Patients with clinically affected relatives were considered as having familial disease. Criteria for diagnosis of HCM in relatives included a maximal wall thickness on echocardiography >13mm or the presence of major ECG criteria: LVH (assessed by a Romhilt-Estes score ≥4) and/or pathological Q waves, in the absence of any other possible causal condition. When HCM could not be clinically diagnosed in relatives (absence of family history and no phenotypic expression on ECG or echocardiogram), HCM was considered to be sporadic or inconclusive (if ≤2 relatives were available for study). In addition, if the genetic testing detected a sequence variant considered pathogenic in the index patient and also in other family members (even with a negative phenotype), the disease was classified as familial.
Molecular analysisAfter informed consent for genotyping was obtained, systematic mutation screening was performed in all patients in the following sarcomeric genes: MYBPC3, MYH7, TNNT2, TNNI3 and the regulatory myosin light chain (MYL2) gene. Genomic DNA was extracted from 10ml of peripheral whole blood using standard techniques. Intronic sets of oligonucleotide primers were designed according to the published genomic sequences of the genes. All exon–intron boundaries were amplified. Mutation screening was performed by PCR followed by direct sequencing on an ABI 3100 DNA sequencer, using the BigDye terminator mix (Applied Biosystems, Lisbon, Portugal).
The first two genes screened were MYBPC3 and MYH7, the two most frequently described as involved in HCM. But even when a mutation was found in one or both of these genes, screening in the other genes proceeded due to the reported frequency of multiple mutations (in the same or in different genes) and their possible impact on prognosis.9 When a previously unreported sequence variant (mutation databases: http://genepath.med.harvard.edu and http://www.hgmd.org/) was identified in the index patient and was considered a possible pathogenic mutation, available first-degree relatives were tested for the identified variant in order to analyze cosegregation with the disease in the family. Except for nonsense and frameshift mutations due to short insertions or deletions and splice site mutations, a missense variant was considered pathogenic when: (1) it affected an amino acid conserved among species; (2) it was absent in another 100 samples; (3) it cosegregated with the disease in the family. For analysis of amino acid conservation we used the in silico sequence alignment program Homologene (http://www.ncbi.nlm.nih.gov/homologene). Further, to estimate the functional consequences of the novel identified missense mutations we analyzed these variations with the evolutionary model program SNPs&GO (http://snps-and-go.biocomp.unibo.it).19 This in-silicon program takes into account information derived from protein sequence, protein sequence profile and protein function. For each predicted effect the program determines a reliability index (RI) of the prediction, from 0 (unreliable) to 10 (reliable).
The mutation nomenclature was based on protein reference sequences: MIM NP_000247 for MYBPC3, MIM NP_000248.1 for MYH7, MIM NP_001001430.1 for TNNT2, MIM NP_000354 for TNNI3 and NP_000423.2 for MYL2.
If no mutation was identified in the five genes studied, index patients with sporadic or inconclusive disease were additionally tested for Anderson-Fabry disease (genetic screening of the GLA gene) considering that, although rare, this disease may be expressed in an entirely or predominantly cardiac phenotype.
Statistical analysisClinical and genetic data were presented as total number or as frequency and percentage. Mean values±standard deviation are expressed when comparing groups. Differences between groups were compared using the Student's t-test or analysis of variance. Fisher's exact test was used for comparing proportions. A p value of <0.05 was considered significant for all analyses.
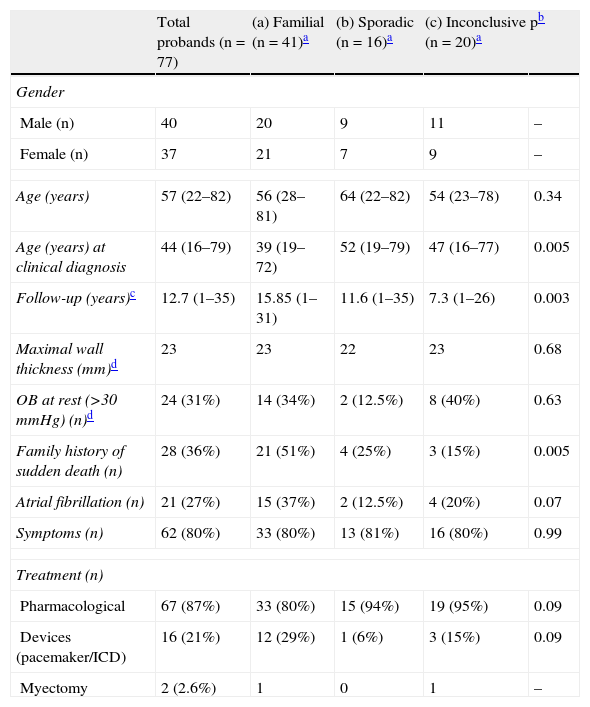
ResultsStudy populationThe clinical data of the cohort under study are shown in Table 1.
Clinical features of HCM index patients (n=77). Comparison of familial, sporadic and inconclusive disease.
| Total probands (n=77) | (a) Familial (n=41)a | (b) Sporadic (n=16)a | (c) Inconclusive (n=20)a | pb | |
| Gender | |||||
| Male (n) | 40 | 20 | 9 | 11 | – |
| Female (n) | 37 | 21 | 7 | 9 | – |
| Age (years) | 57 (22–82) | 56 (28–81) | 64 (22–82) | 54 (23–78) | 0.34 |
| Age (years) at clinical diagnosis | 44 (16–79) | 39 (19–72) | 52 (19–79) | 47 (16–77) | 0.005 |
| Follow-up (years)c | 12.7 (1–35) | 15.85 (1–31) | 11.6 (1–35) | 7.3 (1–26) | 0.003 |
| Maximal wall thickness (mm)d | 23 | 23 | 22 | 23 | 0.68 |
| OB at rest (>30mmHg) (n)d | 24 (31%) | 14 (34%) | 2 (12.5%) | 8 (40%) | 0.63 |
| Family history of sudden death (n) | 28 (36%) | 21 (51%) | 4 (25%) | 3 (15%) | 0.005 |
| Atrial fibrillation (n) | 21 (27%) | 15 (37%) | 2 (12.5%) | 4 (20%) | 0.07 |
| Symptoms (n) | 62 (80%) | 33 (80%) | 13 (81%) | 16 (80%) | 0.99 |
| Treatment (n) | |||||
| Pharmacological | 67 (87%) | 33 (80%) | 15 (94%) | 19 (95%) | 0.09 |
| Devices (pacemaker/ICD) | 16 (21%) | 12 (29%) | 1 (6%) | 3 (15%) | 0.09 |
| Myectomy | 2 (2.6%) | 1 | 0 | 1 | – |
HCM: hypertrophic cardiomyopathy; OB: obstruction (outflow gradient) at rest measured by Doppler echocardiography; ICD: implantable cardioverter-defibrillator.
At the time of genetic testing, all but five patients had predominantly septal hypertrophy, although in many patients other ventricular walls were also involved, with 35 patients showing diffuse LVH. In five patients hypertrophy was predominantly or exclusively apical. Most index patients also had major ECG criteria for HCM.
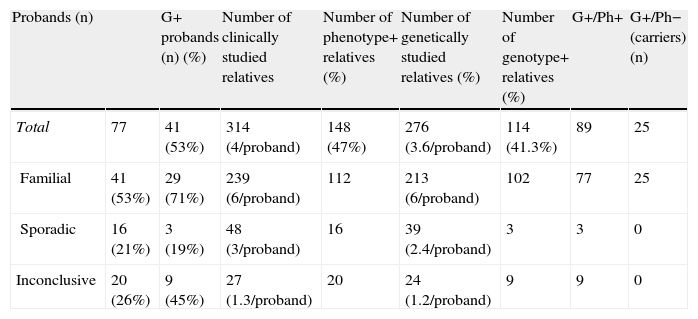
On the basis of phenotype/genotype data (on average four individuals studied in each pedigree), disease was confirmed as familial in 41 cases (53%) and considered sporadic in 16 (21%). In 20 (26%) index patients no conclusions were possible (small families or no relatives available for study) (Table 2).
Analysis of clinical and genetic studies performed in probands and their relatives.
| Probands (n) | G+ probands (n) (%) | Number of clinically studied relatives | Number of phenotype+ relatives (%) | Number of genetically studied relatives (%) | Number of genotype+ relatives (%) | G+/Ph+ | G+/Ph− (carriers) (n) | |
| Total | 77 | 41 (53%) | 314 (4/proband) | 148 (47%) | 276 (3.6/proband) | 114 (41.3%) | 89 | 25 |
| Familial | 41 (53%) | 29 (71%) | 239 (6/proband) | 112 | 213 (6/proband) | 102 | 77 | 25 |
| Sporadic | 16 (21%) | 3 (19%) | 48 (3/proband) | 16 | 39 (2.4/proband) | 3 | 3 | 0 |
| Inconclusive | 20 (26%) | 9 (45%) | 27 (1.3/proband) | 20 | 24 (1.2/proband) | 9 | 9 | 0 |
G+: patients with identified mutation; Ph+: patients with HCM phenotype; Ph−: patients with no HCM phenotype.
Cross-comparison between familial and sporadic or inconclusive HCM showed no differences in probands regarding degree of ventricular hypertrophy, presence of obstruction or symptoms or need for pharmacological treatment or device implantation. But familial cases had been diagnosed at a younger age and more frequently had a family history of premature sudden death (Table 1).
No patient had clinical involvement other than of the heart. Muscle enzymes and renal function were also normal in all index patients.
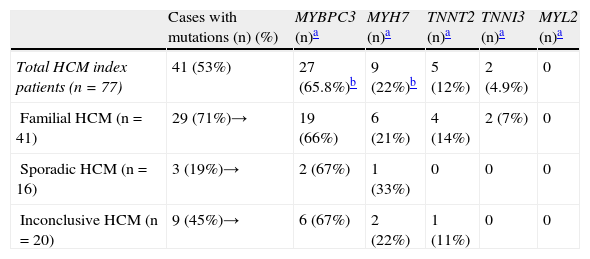
Sarcomeric mutationsSarcomeric mutations were identified in 41 (53%) out of 77 index patients (Tables 2 and 3), more frequently in familial HCM with 29 out of 41 patients (71%) having a positive genotype (genotype+).
HCM probands: genotype status (77 index patients).
| Cases with mutations (n) (%) | MYBPC3 (n)a | MYH7 (n)a | TNNT2 (n)a | TNNI3 (n)a | MYL2 (n)a | |
| Total HCM index patients (n=77) | 41 (53%) | 27 (65.8%)b | 9 (22%)b | 5 (12%) | 2 (4.9%) | 0 |
| Familial HCM (n=41) | 29 (71%)→ | 19 (66%) | 6 (21%) | 4 (14%) | 2 (7%) | 0 |
| Sporadic HCM (n=16) | 3 (19%)→ | 2 (67%) | 1 (33%) | 0 | 0 | 0 |
| Inconclusive HCM (n=20) | 9 (45%)→ | 6 (67%) | 2 (22%) | 1 (11%) | 0 | 0 |
HCM: hypertrophic cardiomyopathy.
Considering the population with identified mutations, MYBPC3 was the most commonly involved gene, with 27 out of 41 patients harboring mutations in this gene (66% of genotype+ patients). Mutations in MYH7 were identified in nine patients (22%), TNNT2 mutations in five patients (12%) and TNNI3 mutations in two patients. In total, mutations in MYBPC3 and/or MYH7 were found in 83% (34 out of 41 patients) with a positive genotype. Double or compound heterozygosity was identified in three patients (7%): two patients had one mutation in MYH7 and another in MYBPC3, and one had two mutations in MYBPC3 (Tables 4–6). No mutations were found in MYL2.
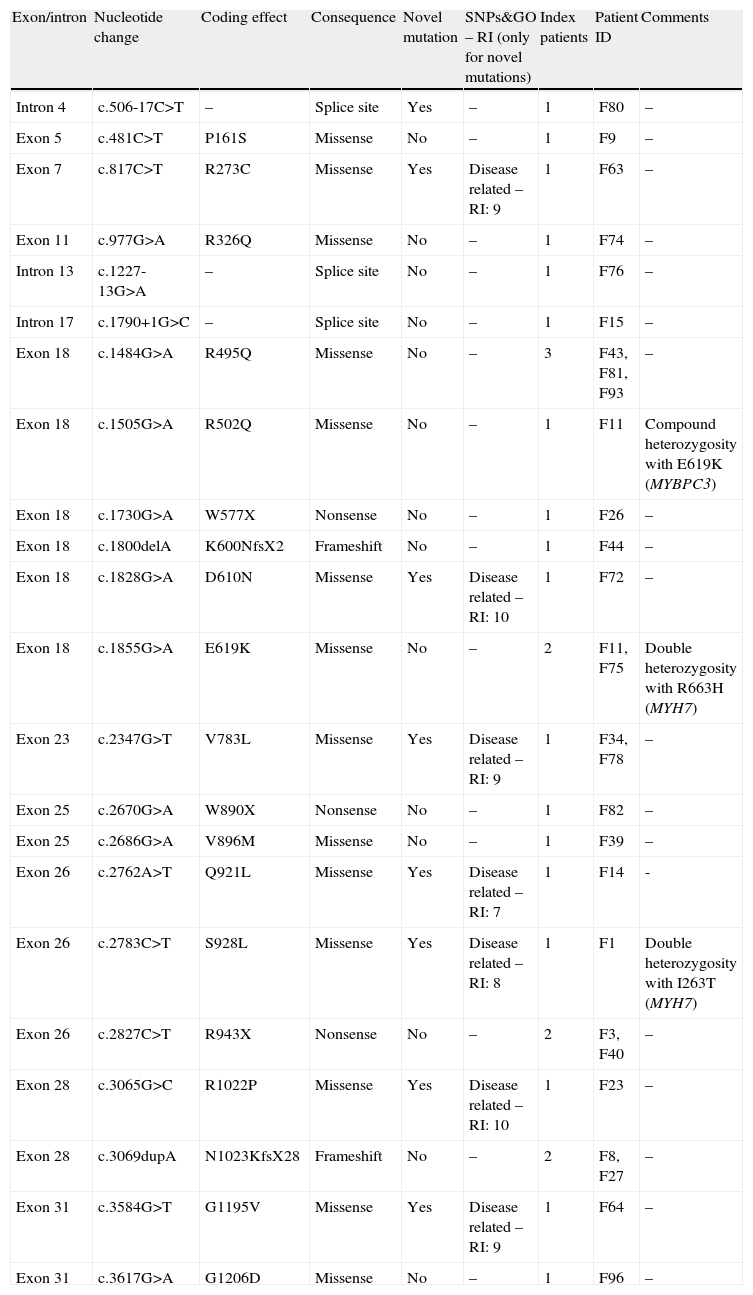
Spectrum of mutations in MYBPC3 gene (27 index patients).
| Exon/intron | Nucleotide change | Coding effect | Consequence | Novel mutation | SNPs&GO – RI (only for novel mutations) | Index patients | Patient ID | Comments |
| Intron 4 | c.506-17C>T | – | Splice site | Yes | – | 1 | F80 | – |
| Exon 5 | c.481C>T | P161S | Missense | No | – | 1 | F9 | – |
| Exon 7 | c.817C>T | R273C | Missense | Yes | Disease related – RI: 9 | 1 | F63 | – |
| Exon 11 | c.977G>A | R326Q | Missense | No | – | 1 | F74 | – |
| Intron 13 | c.1227-13G>A | – | Splice site | No | – | 1 | F76 | – |
| Intron 17 | c.1790+1G>C | – | Splice site | No | – | 1 | F15 | – |
| Exon 18 | c.1484G>A | R495Q | Missense | No | – | 3 | F43, F81, F93 | – |
| Exon 18 | c.1505G>A | R502Q | Missense | No | – | 1 | F11 | Compound heterozygosity with E619K (MYBPC3) |
| Exon 18 | c.1730G>A | W577X | Nonsense | No | – | 1 | F26 | – |
| Exon 18 | c.1800delA | K600NfsX2 | Frameshift | No | – | 1 | F44 | – |
| Exon 18 | c.1828G>A | D610N | Missense | Yes | Disease related – RI: 10 | 1 | F72 | – |
| Exon 18 | c.1855G>A | E619K | Missense | No | – | 2 | F11, F75 | Double heterozygosity with R663H (MYH7) |
| Exon 23 | c.2347G>T | V783L | Missense | Yes | Disease related – RI: 9 | 1 | F34, F78 | – |
| Exon 25 | c.2670G>A | W890X | Nonsense | No | – | 1 | F82 | – |
| Exon 25 | c.2686G>A | V896M | Missense | No | – | 1 | F39 | – |
| Exon 26 | c.2762A>T | Q921L | Missense | Yes | Disease related – RI: 7 | 1 | F14 | - |
| Exon 26 | c.2783C>T | S928L | Missense | Yes | Disease related – RI: 8 | 1 | F1 | Double heterozygosity with I263T (MYH7) |
| Exon 26 | c.2827C>T | R943X | Nonsense | No | – | 2 | F3, F40 | – |
| Exon 28 | c.3065G>C | R1022P | Missense | Yes | Disease related – RI: 10 | 1 | F23 | – |
| Exon 28 | c.3069dupA | N1023KfsX28 | Frameshift | No | – | 2 | F8, F27 | – |
| Exon 31 | c.3584G>T | G1195V | Missense | Yes | Disease related – RI: 9 | 1 | F64 | – |
| Exon 31 | c.3617G>A | G1206D | Missense | No | – | 1 | F96 | – |
ID: patient identification; SNP&GO – RI: reliability index of the predicted effect (see text for details).
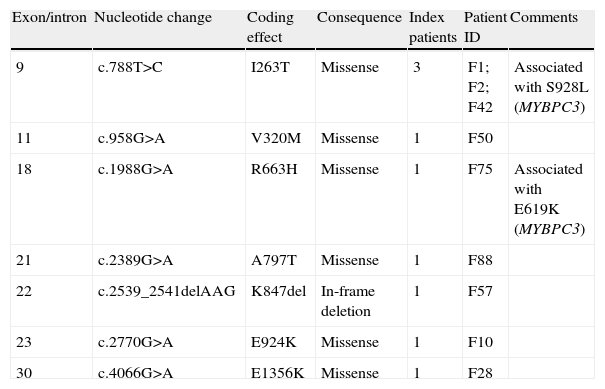
Spectrum of mutations in MYH7 gene (9 index patients).
| Exon/intron | Nucleotide change | Coding effect | Consequence | Index patients | Patient ID | Comments |
| 9 | c.788T>C | I263T | Missense | 3 | F1; F2; F42 | Associated with S928L (MYBPC3) |
| 11 | c.958G>A | V320M | Missense | 1 | F50 | |
| 18 | c.1988G>A | R663H | Missense | 1 | F75 | Associated with E619K (MYBPC3) |
| 21 | c.2389G>A | A797T | Missense | 1 | F88 | |
| 22 | c.2539_2541delAAG | K847del | In-frame deletion | 1 | F57 | |
| 23 | c.2770G>A | E924K | Missense | 1 | F10 | |
| 30 | c.4066G>A | E1356K | Missense | 1 | F28 |
ID: patient identification.
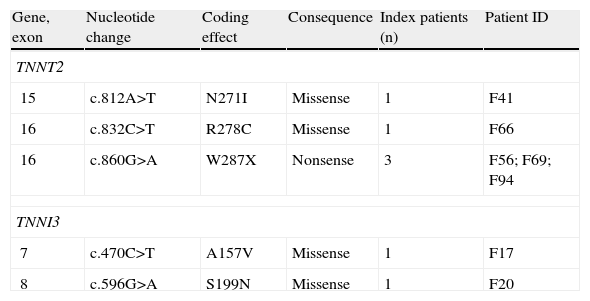
Spectrum of mutations in TNNT2 and TNNI3 genes.
| Gene, exon | Nucleotide change | Coding effect | Consequence | Index patients (n) | Patient ID |
| TNNT2 | |||||
| 15 | c.812A>T | N271I | Missense | 1 | F41 |
| 16 | c.832C>T | R278C | Missense | 1 | F66 |
| 16 | c.860G>A | W287X | Nonsense | 3 | F56; F69; F94 |
| TNNI3 | |||||
| 7 | c.470C>T | A157V | Missense | 1 | F17 |
| 8 | c.596G>A | S199N | Missense | 1 | F20 |
ID: patient identification.
No patient with no identified mutations in sarcomeric genes had Anderson-Fabry disease.
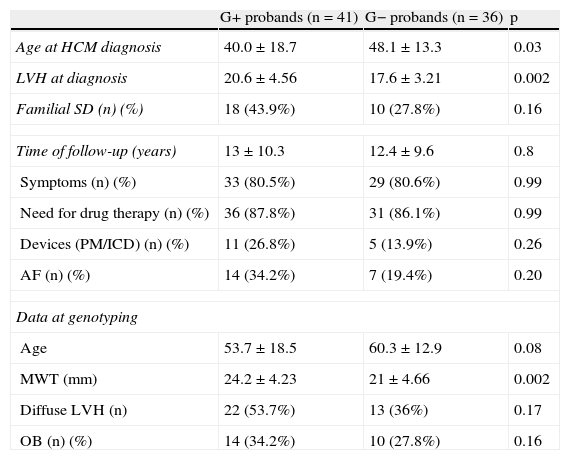
No differences were observed between index patients with or without identified mutations in terms of gender, age, time of follow-up, morbidity (symptoms, atrial fibrillation, hospitalizations during follow-up or need for device implantation) or family history of sudden death. However, probands with identified mutations were younger at the time of diagnosis and had more severe septal hypertrophy (at diagnosis and after follow-up) (Table 7).
Comparison between index patients with and without identified mutations.
| G+ probands (n=41) | G− probands (n=36) | p | |
| Age at HCM diagnosis | 40.0±18.7 | 48.1±13.3 | 0.03 |
| LVH at diagnosis | 20.6±4.56 | 17.6±3.21 | 0.002 |
| Familial SD (n) (%) | 18 (43.9%) | 10 (27.8%) | 0.16 |
| Time of follow-up (years) | 13±10.3 | 12.4±9.6 | 0.8 |
| Symptoms (n) (%) | 33 (80.5%) | 29 (80.6%) | 0.99 |
| Need for drug therapy (n) (%) | 36 (87.8%) | 31 (86.1%) | 0.99 |
| Devices (PM/ICD) (n) (%) | 11 (26.8%) | 5 (13.9%) | 0.26 |
| AF (n) (%) | 14 (34.2%) | 7 (19.4%) | 0.20 |
| Data at genotyping | |||
| Age | 53.7±18.5 | 60.3±12.9 | 0.08 |
| MWT (mm) | 24.2±4.23 | 21±4.66 | 0.002 |
| Diffuse LVH (n) | 22 (53.7%) | 13 (36%) | 0.17 |
| OB (n) (%) | 14 (34.2%) | 10 (27.8%) | 0.16 |
AF: atrial fibrillation; HCM: hypertrophic cardiomyopathy; ICD: implantable cardioverter-defibrillator; genotype-positive (+): index patients with identified mutations; genotype negative (−): index patients without identified mutations; LVH: left ventricular hypertrophy; MWT: maximal wall thicknesss; OB: presence of obstruction; PM: pacemaker; SD: sudden death.
The spectrum of identified mutations is described in Tables 4–6.
MYBPC3Twenty-two different mutations were identified in MYBPC3 in 27 probands (Table 4). Fourteen mutations were missense (seven novel). The other mutations included three nonsense, two frameshift and three intronic mutations (one also novel). The novel and multiple mutations are discussed separately (see below).
Fourteen different mutations already reported as pathogenic were identified in 18 unrelated probands. Most mutations were private, but identical mutations were detected in four unrelated pedigrees. The R495Q mutation was found in three unrelated probands (F43, F81 and F93), two with familial HCM and one with inconclusive HCM. The R943X mutation was identified in two probands (F3 – familial HCM; F40 – inconclusive). The N1023KfsX28 mutation was found in two unrelated probands (F8 and F27, both with familial HCM), and the E619K mutation was found in two unrelated probands (F11 and F75, both with familial HCM). The latter two probands demonstrated complex genetic status, with compound heterozygosity in F11 (two different mutations in MYBPC3) and double heterozygosity in F75 (one mutation in MYBPC3 and one in MYH7) – see “Multiple mutations” below.
For cosegregation analysis in the overall population of probands with identified mutations in the MYBPC3 gene, 105 additional relatives were clinically screened and 82 were genetically tested. Familial disease was diagnosed in 19 out of 27 pedigrees. The other eight probands were considered to have either sporadic or inconclusive disease.
MYH7In this gene, seven different mutations were identified in nine unrelated probands (Table 5). All seven had already been described as associated with HCM.5,6,20–29 The missense mutation I263T was identified in three unrelated patients. In two, disease is familial (F1 and F2), with 19 individuals studied in each family and cosegregation with the disease in both. Besides the I263T mutation, another mutation (S928L in MYBPC3, novel) was also identified in the first family (F1, double heterozygosity) with unknown functional consequences. The other proband with the I263T mutation (F42) was the only individual studied in his pedigree and was thus considered to have an inconclusive type of disease.
The other six mutations were identified in one proband each. Four of these missense mutations are associated with familial HCM – V320M, R663H, E924K, and E1356K – with 17 relatives studied in total and demonstrated cosegregation with the HCM phenotype in each family. The R663H mutation (familial disease) coexisted with another mutation (E619K) in MYBPC3 (double heterozygosity).
Two mutations were considered to be associated with either sporadic HCM (K847del) or inconclusive disease (A797T) due to the fact that few relatives were available for study.
TNNT2Three different mutations were identified in five probands (Table 6). All mutations were previously reported as pathogenic.5,30–32
The N271I missense mutation was identified in one patient and proved to be familial (13 relatives studied). The R278C missense mutation was identified in a small family with a mother and daughter both affected. There was a family history of premature (aged <50 years) sudden death in a first-degree relative. The nonsense W287X mutation was identified in three unrelated probands, two of them with familial disease (cosegregation with the disease in each family). In the case of the other proband, no other relatives were available for study and the familial character was considered inconclusive.
TNNI3Two different missense mutations – A157V and S199N – were identified in this gene (Table 6), in two unrelated patients, both with familial disease and with more than 15 years of follow-up. Both mutations had been reported as pathogenic.5,33,34
Multiple mutationsIn three probands (4% of the overall population and 7% of genotype-positive patients), two mutations were found (Tables 4 and 5). All three patients have familial HCM. One carries two mutations in MYBPC3 (compound heterozygosity): R502Q and E619K – both previously described. The other two patients showed double heterozygosity: E619K in MYBPC3 with R663H in MYH7 in one patient (both mutations previously described); and I263T in MYH7 and S928L in MYBPC3 in the other. The latter mutation in MYBPC3 is novel and although its RI is 8 its functional consequences are still unknown.
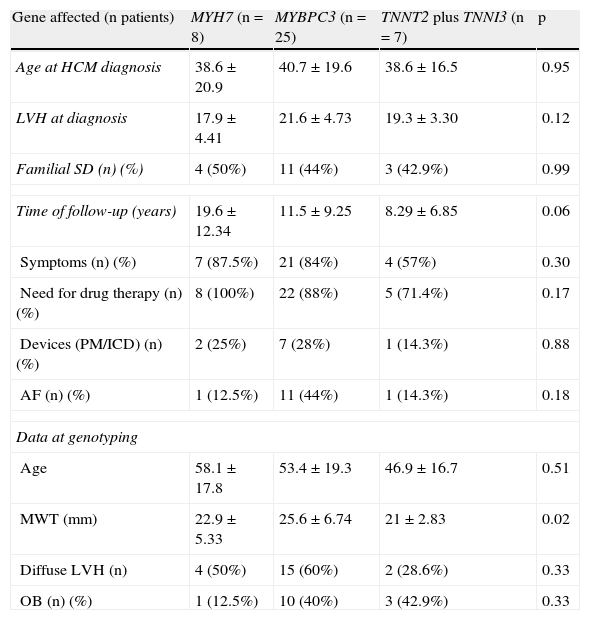
A comparison was performed regarding clinical characteristics between index patients with single mutations in the MYH7, MYBPC3 and TNNT2 plus TNNI3 genes (Table 8). Patients with double mutations were excluded from this analysis. No differences were observed regarding age, degree of LVH at time of diagnosis, length of follow-up, morbidity or family history of sudden death. However, an association was found between the affected gene and maximal wall thickness, with higher values in patients with identified mutations in MYBPC3 than in patients with mutations in the other genes (after the same follow-up period).
Comparison between index patients with mutations in different genes.
| Gene affected (n patients) | MYH7 (n=8) | MYBPC3 (n=25) | TNNT2 plus TNNI3 (n=7) | p |
| Age at HCM diagnosis | 38.6±20.9 | 40.7±19.6 | 38.6±16.5 | 0.95 |
| LVH at diagnosis | 17.9±4.41 | 21.6±4.73 | 19.3±3.30 | 0.12 |
| Familial SD (n) (%) | 4 (50%) | 11 (44%) | 3 (42.9%) | 0.99 |
| Time of follow-up (years) | 19.6±12.34 | 11.5±9.25 | 8.29±6.85 | 0.06 |
| Symptoms (n) (%) | 7 (87.5%) | 21 (84%) | 4 (57%) | 0.30 |
| Need for drug therapy (n) (%) | 8 (100%) | 22 (88%) | 5 (71.4%) | 0.17 |
| Devices (PM/ICD) (n) (%) | 2 (25%) | 7 (28%) | 1 (14.3%) | 0.88 |
| AF (n) (%) | 1 (12.5%) | 11 (44%) | 1 (14.3%) | 0.18 |
| Data at genotyping | ||||
| Age | 58.1±17.8 | 53.4±19.3 | 46.9±16.7 | 0.51 |
| MWT (mm) | 22.9±5.33 | 25.6±6.74 | 21±2.83 | 0.02 |
| Diffuse LVH (n) | 4 (50%) | 15 (60%) | 2 (28.6%) | 0.33 |
| OB (n) (%) | 1 (12.5%) | 10 (40%) | 3 (42.9%) | 0.33 |
Patients with more than one mutation were excluded from the comparison. AF: atrial fibrillation; HCM: hypertrophic cardiomyopathy; ICD: implantable cardioverter-defibrillator; LVH: left ventricular hypertrophy; MWT: maximal wall thickness; OB: presence of obstruction; PM: pacemaker; SD: sudden death.
Eight novel variants were identified in the study population (Table 4), all in the MYBPC3 gene.
Seven of the mutations were missense. All were detected in coding regions of the gene. The affected amino acids are conserved in various species and the mutations were not detected in any others of 100 samples. Their effects predicted by the SNPs&GO program led them to be considered as disease-related, with an RI of 7–10. An RI of 7 or 8 was found for only one mutation each (see Table 4 and below), the other five novel mutations having an RI of 9 or 10. All of these novel mutations were found in only one proband, except V783L, which was identified in two index patients (one with familial disease).
The seven index patients with these novel missense mutations were followed for a mean of 15 (1–30) years before genotyping and had a mean maximal wall thickness of 26 (21–32)mm at the time of genotyping. Six of them (86%) showed considerable HCM-related morbidity during follow-up – hospitalization or need for devices or surgery (myectomy or mitral prosthesis) – and three have a family history of premature sudden death. In total, 40 relatives were studied for cosegregation analysis. From these cosegregation studies, five of the seven novel mutations were associated with familial disease. Nevertheless, as mentioned above, the pathogenicity of the S928L mutation (with an RI of 8) is unclear, as it occurred together with another disease-related mutation in the pedigree.
Concerning the other two of the seven novel missense mutations, Q921L was identified in a proband considered to have sporadic HCM (RI for this mutation was 7). The G1195V mutation (RI of 9) was detected in one proband with HCM and with a history of premature sudden cardiac death in a first-degree relative. No further relatives are available for study, and thus the sporadic or familial character of HCM in these cases was considered inconclusive.
A novel intronic variant (c.506-17C>T) in MYBPC3 was also identified in a patient with obstructive HCM and severe LVH. Familial studies are proceeding, but the significance of this new variant is for the moment unknown.
Additional results on familial, sporadic or inconclusive diseaseFamilial HCMConcerning the 41 unrelated probands with familial HCM, mutations were identified in 29 (71%). Clinical screening was performed in 239 relatives and genetic screening in 213 (on average six individuals were studied in each family) (Table 2).
Clinically, 112 individuals out of 239 (47%) had HCM; genetically, 102 out of 213 (47%) had identified mutations. It should be noted that not all clinically affected individuals were genetically screened but those who were had a positive genotype (77 relatives genotype+/phenotype+). Additionally, 25 carriers were identified.
Sporadic HCMSixteen index patients were considered to have sporadic disease (21% of the study cohort). A mutation was identified in three (7% of patients with a positive genotype). The novel variant Q921L in MYBPC3 was found in a patient with diffuse LVH and a MWT of 31mm (septal), subaortic obstruction and severe heart failure with preserved systolic function (no living first-degree relatives). The c.1790+1G>C mutation (not novel) was also identified in MYBPC3 in a young woman with LVH and clinically normal first-degree relatives. The previously described K847E mutation in MYH7 was found in one patient. A total of 48 relatives of pedigrees with sporadic HCM were clinically screened (about three in each family) and in 39 genetic testing was also performed. None had a clinical phenotype or a positive genetic test.
Inconclusive HCMIn 20 index patients, no conclusion regarding the familial or sporadic character of HCM could be drawn due to the small number of relatives studied. However, a mutation was identified in nine of these 20 index patients (22% of the population with a positive genotype). Of these, six mutations were already described as pathogenic and three were novel.
Concerning the six previously described mutations (MYBPC3: R943ter, R326Q, R495Q; MYH7: I263T, A797T; TNNT2: Trp287X), four were also identified as causing familial HCM in other probands of our population: R943X (MYBPC3) in one pedigree; R495Q (MYBPC3) in two pedigrees; I263T (MYH7) in two pedigrees (see above); and W287X (TNNT2) in two other pedigrees. Thus four cases of inconclusive HCM might be in fact familial, although an insufficient number of relatives were available for study. The other two previously reported mutations – R326Q (MYBPC3) and A797T (MYH7) – were only found in two unrelated probands and were also considered to be inconclusive cases.
Of the three novel mutations in MYBPC3 in patients with inconclusive disease, the G1195V mutation was detected in only one patient (see above) with no relatives available for study. The V783L mutation – with an RI of 9 – was first found in one proband with disease diagnosed 36 years previously and followed since then. However, the same mutation was also identified in a case of familial HCM in our cohort, with several relatives studied and cosegregation demonstrated with the disease in that family. The novel variant in intron 4 (c.506-17C>T) was identified in one proband. The pathogenicity of this variant is uncertain.
DiscussionThe present study reports the frequency and profile of single and multiple mutations in a Portuguese cohort of unrelated HCM index patients, most with an established diagnosis for many years (mean follow-up 12.7 years). The entire coding regions of five sarcomeric genes were screened for alterations in order to identify all described and as yet unreported mutations. Our results demonstrated an overall mutation detection rate of 53%. This is in agreement with previously reported data in other population studies.4–6,8,9,12,15 All mutations in our cohort were identified in the four genes most commonly associated with HCM (MYBPC3, MYH7, TNNT2 and TNNI3). Familial HCM showed the highest rate of mutations, comprising 71% of the population with a positive genotype.
In a recent study that included only HCM index patients genotyped for these four genes,15 a higher rate of mutations in familial disease (64%) was found in a French population compared to sporadic (29%) or inconclusive disease (46%). These data corroborate previously published studies in the French population showing a 70% frequency of sarcomeric mutations in familial HCM.5 In our cohort, only 7% (three out of 16 patients) of sporadic HCM and 22% of patients (9 out of 20) with inconclusive disease had an identified mutation.
Differences concerning sporadic or inconclusive disease may be explained by differences in the populations studied, as is always the case with HCM. The number of relatives available for studying cosegregation with the disease in each family is highly variable in different published series, as is the information on a diagnosis of HCM in relatives. Additionally, a different number of genes are screened in each population studied. Studies that include predominantly familial HCM have a high rate of genotype-positive patients,12 as is the case with our study. Sporadic HCM was found in only 21% of our cohort; it is less frequent than familial HCM and is diagnosed later.
Our findings show that many of the inconclusive HCM cases are in fact familial. The study of as many relatives as possible in each family should be the goal in HCM genetic population studies.
In 36 patients (46%) – including 29% of those with familial HCM – no mutations were identified in the five genes screened.
We are aware that screening only five sarcomeric genes may have missed some genotype cases in other genes, although most myofilament mutations associated with HCM are usually identified in the four genes mutated in our population. No mutations were found in MYL2 in our population, and the reported mean frequency of mutations in TPM1 (encoding α-tropomyosin), MYL3 (myosin essential light chain) and ACTC (encoding α-actin) is very low, 1.21%, 0.11% and 0.49%, respectively.12 It is commonly thought that since HCM is a genetically heterogeneous disease, patients with HCM and no identified mutations in the known myofilament coding genes may in fact have mutations in unexplored regions of these genes. Alternatively mutations may exist in novel myofilament encoding genes (like titin, muscle LIM protein, telethonin, myozenin 2 or vinculin) or in other HCM-susceptibility genes that were not screened in our cohort.
Some differences were observed in our population between patients with and without myofilament identified mutations. Patients with a positive genotype were younger at the time of diagnosis and showed more severe septal hypertrophy (at diagnosis and after the same follow-up time). The implications of these findings need further study.
Mutations in MYBPC3 were the most prevalent cause of HCM in our cohort, with 35% of probands affected. The frequency of mutations in MYBPC3 has been reported as ranging from 14% to 26% in HCM.12,15 A recent study of an Italian cohort including 203 unrelated HCM patients9 found that 34% had mutations in MYBPC3, a similar frequency to ours. Although defining the pathogenicity of mutations in MYBPC3 (especially for missense mutations) is particularly challenging, the criteria applied in our study for the novel variants identified support the idea that most of the missense variants identified are indeed pathogenic and their effects are not overestimated.
The prevalence of patients with mutations in the MYH7 gene was 11.7%. Findings in other studies ranged from 2.8% to 25%.12 The number of patients with mutations in MYH7 in our study is unlikely to be underestimated as the entire coding sequence of the gene was analyzed. Mutations in this gene and in MYBPC3 were found in 87% of genotype-positive patients, a frequency close to that reported in other studies.5,9,15
The TNNT2 gene was affected in 6.5% of HCM probands (12% of genotype-positive patients). Mutations in this gene are the most commonly involved in thin filament-HCM, with reported values for affected probands ranging from 0% to 6.67%.22,35 Mutations in the TNNI3 gene were also found in 2.6% of HCM patients (4.9% of those with identified mutations), in agreement with previously reported rates (ranging from 0% to 6.5%).12,15
Multiple mutations were detected in 7% of index patients with a positive genotype. This finding is in line with previously published studies4,5,7,9 and indicates that once a single sarcomeric mutation is identified in a patient a continued search for other mutations is necessary, at least in MYBPC3 and MYH7, the two genes most commonly associated with sarcomeric HCM.
Comparison of clinical data of patients with single mutations in different genes showed no significant differences except for a higher degree of hypertrophy after follow-up in patients with MYBPC3 gene mutations. The possible interpretations of this finding are beyond the scope of this study, since the rate of different private mutations in this gene was very high and the specific functional effect of each mutation on disease expression is unknown. Furthermore, it is not the purpose of this study to establish associations between specific genotypes and phenotypes in HCM or to put forward considerations about individual prognosis.
A genetic diagnosis establishing a causal relationship with HCM is important for several reasons. First, it identifies affected individuals (excluding other less prevalent genetic causes of cardiac hypertrophy with different outcomes). Second, it enables identification of family members with a positive genotype at risk of developing HCM who require rigorous and serial follow-up for risk stratification. Third, knowledge of the genetic cause of HCM not only may in the near future help direct treatment strategies in already clinically affected patients36 but also, within families with different mutations, the identification of carriers (in a preclinical state) may enable preventive interventions to limit disease development, a possibility that is already under study (http://clinicaltrials.gov/ct2/show/record/NCT00319982). Additionally genetic study should be performed to reassure genotype-negative family members.
ConclusionsThe molecular screening of five sarcomeric genes associated with HCM in a Portuguese population enabled a genetic diagnosis in 53% of a consecutive cohort of unrelated patients. The highest yield was observed in familial HCM (71%). The most commonly mutated gene was MYBPC3, but since mutations in this gene and in MYH7 account for most genotype-positive patients and more than one mutation was found in 4% of HCM patients, both genes should be systematically screened as a first genetic approach by testing the entire coding regions of these genes. Private mutations were the rule and novel mutations were a frequent finding, illustrating the difficulty of making genotype-phenotype correlations in HCM on an individual basis for each mutation. Independently of the gene affected, genotype-positive patients were younger at the time of diagnosis and had more severe hypertrophy than patients with a negative genotype, although the significance of these findings and their impact on prognosis needs further evaluation. The identification of a positive genotype in probands led to confirmation of the disease in a substantial number of relatives, including 25 carriers (genetically positive, preclinical state) with implications for follow-up, risk stratification and genetic counseling.
Conflicts of interestThe authors have no conflicts of interest to declare.
