






Inhibition of platelet aggregation appears two hours after the first dose of clopidogrel, becomes significant after the second dose, and progresses to a steady-state value of 55% by day seven. Low response to clopidogrel has been associated with increased risk of stent thrombosis and ischemic events, particularly in the context of stable heart disease treated by percutaneous coronary intervention.
ObjectiveTo stratify medium-term prognosis of an acute coronary syndrome (ACS) population by platelet aggregation.
MethodsWe performed a prospective longitudinal study of 70 patients admitted for an ACS between May and August 2009. Platelet function was assessed by ADP-induced platelet aggregation using a commercially available kit (Multiplate® analyzer) at discharge. The primary endpoint was a combined outcome of mortality, non-fatal myocardial infarction, or unstable angina, with a median follow-up of 136.0 (79.0–188.0) days.
ResultsThe median value of platelet aggregation was 16.0U (11.0–22.5U) with a maximum of 41.0U and a minimum of 4.0U (normal value according to the manufacturer: 53–122U). After ROC curve analysis with respect to the combined endpoint (AUC 0.72), we concluded that a value of 18.5U conferred a sensitivity of 75.0% and a specificity of 68% to that result. We therefore created two groups based on that level: group A – platelet aggregation <18.5U, n=44; and group B – platelet aggregation ≥18.5U, n=26. The groups were similar with respect to demographic data (age 60.5 [49.0–65.0] vs. 62.0 [49.0–65.0] years, p=0.21), previous cardiovascular history, and admission diagnosis. There were no associations between left ventricular ejection fraction, GRACE risk score, or length of hospital stay and platelet aggregation. The groups were also similar with respect to antiplatelet, anticoagulant, proton pump inhibitor (63.6 vs. 46.2%, p=0.15) and statin therapy. The variability in platelets and hemoglobin was also similar between groups. Combined event-free survival was higher in group A (96.0 vs. 76.7%, log-rank p<0.01). Platelet aggregation higher than 18.5U was an independent predictor of the combined event (HR 6.75, 95% CI 1.38–32.90, p=0.02).
ConclusionIn our ACS population platelet aggregation at discharge was a predictor of medium-term prognosis.
A inibição da agregação plaquetar pelo clopidogrel surge nas primeiras 2 horas após início da toma do fármaco e atinge um valor estável de 55% pela sétima toma. Uma fraca resposta ao clopidogrel está associada a um agravamento do prognóstico isquémico, particularmente no contexto de doença coronária estável.
ObjetivoEstratificação prognóstica no contexto de uma síndrome coronária aguda (SCA) pela agregação plaquetar.
População e métodosEstudo prospetivo, longitudinal, de 70 doentes admitidos por uma SCA entre maio e agosto de 2009. A função plaquetar foi determinada pela agregação plaquetar com o ADP, com o recurso a um kit comercial (Multiplate®) à data da alta. O endpoint primário do estudo foi um resultado combinado de morte, enfarte agudo do miocárdio não fatal, e angina instável – o tempo mediano de seguimento foi de 136,0 (79,0-188,0) dias.
ResultadosO resultado mediano da agregação plaquetar foi de 16,0 U (11,0-22,5 U) com um máximo de 41,0 U e mínimo de 4,0 U (valor normal de 53-122 U). Após análise da curva ROC (AUC 0,72), foi concluído que um valor de corte de 18,5 U apresentava uma sensibilidade de 75% e especificidade de 68% para o resultado primário. Foram criados 2 grupos baseados nesse valor: grupo A – agregação pelo ADP<18,5 U, n=44; grupo B – agregação pelo ADP ≥ 18,5 U, n=26. Os grupos foram semelhantes relativamente a dados demográficos (idade: 60,5 [49,0-65,0] versus 62,0 [49,0-65,0] anos, p=0,21), história cardiovascular prévia, e diagnóstico de admissão. Não existiram associações entre a fração de ejeção do ventrículo esquerdo, o score de GRACE, a duração do internamento hospitalar, e a agregação plaquetar. Os grupos foram também homogéneos relativamente à terapêutica antiagregante, anticoagulante, uso de inibidores da bomba de protões (63,6 versus 46,2%, p=0,15) e estatinas. A variação da hemoglobina e das plaquetas durante o internamento foi também semelhante para ambos os grupos. A sobrevida livre de eventos foi superior no grupo A (96,0 versus 76,7%, Log Rank p<0,01). Um valor de agregação plaquetar superior a 18,5 U foi um preditor independente do evento combinado (HR 6,75, IC95% 1,38-32,9, p=0,02).
ConclusãoNa população estudada a agregação plaquetar à data de alta de uma SCA foi um preditor independente de prognóstico.
The ADP-receptor blocker clopidogrel reduces the incidence of recurrent ischemic events in patients with acute coronary syndrome (ACS)1 and after coronary stenting.2 Nevertheless up to 15% of high-risk ACS patients continue to suffer from ischemic events.3
Marked interindividual variability in the extent of platelet inhibition is seen in up to a third of patients treated with clopidogrel, and a recent meta-analysis found an overall prevalence of 21% laboratory-defined clopidogrel non-responsiveness.4
Low response to clopidogrel has been associated with increased risk of stent thrombosis5 and ischemic events, particularly in the context of stable heart disease treated by percutaneous coronary intervention (PCI).6,7
Inhibition of platelet aggregation usually appears two hours after the first dose of clopidogrel, becomes significant after the second, and progresses to a steady-state value of 55% by day seven.8
A new point-of-care assay, multiple electrode aggregometry (MEA), has been developed recently for rapid and standardized assessment of platelet function in whole blood.9 MEA is based on the principle of impedance aggregometry, avoiding the need for blood centrifugation, and can assess platelet function in approximately 10min. MEA, implemented in a device called the Multiplate analyzer (Dynabyte®, Munich, Germany), is capable of detecting the effect of clopidogrel treatment, and the results of MEA correlate well with light transmission aggregometry (LTA).10
In this context we decided to prospectively assess platelet reactivity in an unselected ACS population and to correlate it with subsequent ischemic events.
MethodsStudy design and eligibilityWe performed a prospective longitudinal study of 70 consecutive patients admitted for an ACS who survived hospital stay between May and August 2009.
The local ethics committee approved the research protocol and informed consent was obtained.
To be eligible, patients had to be less than 75 years old and to be discharged on clopidogrel therapy (75mg/day).
Patients were excluded if they were immediately referred for surgery, or if they were included in either of two clinical trials related to antiplatelet therapy – TRA•CER (Trial to Assess the Effects of SCH 530348 in Preventing Heart Attack and Stroke in Patients With Acute Coronary Syndrome11 – ClinicalTrials.gov identifier: NCT00527943) and TRILOGY ACS (A Comparison of Prasugrel and Clopidogrel in Acute Coronary Syndrome Subjects – ClinicalTrials.gov identifier: NCT00699998).12
Myocardial infarction (MI) was defined according to the universal definition of myocardial infarction, as the presence of a positive cardiac biomarker (troponin I) with symptoms of ischemia or ECG changes indicative of new ischemia (ST-segment or T-wave changes or new bundle branch block).13
Regarding ECG data, ST-elevation MI was defined by new-onset ST elevation greater than 2mm for men and 1.5mm for women in V2-V3 leads and greater than 1mm in other leads. Non-ST elevation MI, besides the above laboratorial and clinical criteria, may or may not be associated with ischemic ECG changes (ST depression or T-wave inversion).13
Unstable angina was defined as new onset angina (at least CCS class III), progressive angina, or angina at rest, with or without ischemic ECG changes, and a negative cardiac biomarker assay.14
A 300-mg clopidogrel loading dose was administered in the emergency department for non-ST elevation ACS, and a 600-mg dose for ST-elevation ACS. Subsequent daily and discharge dosage was 75mg per day at 8am. The loading dose for aspirin was 300mg, with a subsequent dosage of 100mg per day at 12am.
The initial population was composed of 162 patients. In-hospital mortality was 6% (10 patients) and 52 patients were excluded because they were aged over 75. During the study period 20 patients were randomized to TRA•CER and four to TRILOGY ACS. Six patients were also immediately referred for surgery.
Platelet function testsWhole blood was obtained from a peripheral vein on the day of discharge at 12am in 4-ml plastic tubes containing the anticoagulant citrate.
The blood samples were kept at room temperature for at least 30min before platelet function testing.
Platelet function in whole blood was measured using the Multiplate analyzer.15,16 Details of this method have been reported elsewhere.10 In brief, the analysis was performed in a single-use test cell, which incorporates two independent impedance sensors. For the present analysis 300μl of saline and 300μl of patient blood (anticoagulated with citrate) were pipetted into the test cell and stirred for 3min. The agonist, 6.4μmol/l of ADP (ADP test; Dynabyte Medical), was added and real-time recording started. The ability of platelets to adhere to the metal sensors was assessed for 6min. Platelet adhesion and aggregation were logged by measuring changes in impedance. The resistance change was converted to arbitrary units (U). The normal reference range for healthy blood donors is 53–122U. The analysis was performed in the clinical pathology laboratory. The instrument and all of the reagents are commercially available and were obtained from the manufacturer (Dynabyte).16
GenotypingGenomic DNA was extracted at hospital discharge from peripheral blood leukocytes.
CYP2C19*1 (wild type) and CYP2C19*2 (681 G>A), two SNPs of the CYPC2C19 gene, were genotyped using a commercially available kit from Seegene®.
The technique employed is based on dual priming oligonucleotide (DPO) polymerase chain reaction (PCR). DPO PCR consists of two separate priming regions joined by a polydeoxyinosine linker. This linker is not involved in priming but rather in delineating the boundary between the two parts of the primer. In the first priming reaction the longer 5′ segment binds to the template DNA, initiating stable annealing. In the second priming reaction the short 3′ segment selectively binds to a target site and determines a target-specific extension, acting as a determiner. Conventional primers have a single priming region and extension may proceed even in the presence of mismatches between a primer and a template. The new methodology used is a fundamental tool for blocking extension of non-specifically primed templates, generating consistently high PCR specificity, as previously described for CYP2C19 SNPs.17
A commercial genomic DNA extraction kit was used in order to obtain 60–100ng of isolated DNA for PCR. Amplification was performed in a final volume of 20μl in accordance with the manufacturer's instructions. It contained 4μl DNA solution, 3μl MOP solution, 10μl 2x Multiplex Master Mix (Seegene, Seoul, Korea) and 3μl of template.
The reaction started after an initial preheating step at 94°C for fifteen min, followed by 35 amplification cycles in the thermal cycler under the following conditions: denaturation at 94°C for 30s, annealing at 63°C for 30s, and extension at 72°C for 30s. Amplification was completed with a final extension step at 72°C for 5min. PCR fluorescence yield for the two different dyes was measured to obtain the allelic discrimination plot and to identify individual genotypes and was presented on a 2-dimensional graph.
Baseline data and patient follow-upStandardized records at admission included demographic, clinical, electrical and laboratory data. Medical therapy, catheterization data, in-hospital events and discharge medication were also recorded.
Median clinical follow-up was 136.0 (79.0–188.0) days after hospital discharge. The information was collected by telephone, from hospital records or at the outpatient clinic. The primary endpoint analyzed was the combined outcome of cardiovascular death, non-fatal myocardial infarction or readmission for unstable angina.
For the present study we prospectively defined low responsiveness to clopidogrel by setting a cutoff at the upper quartile (25%) of MEA measurement.
Statistical analysisReceiver–operator characteristic (ROC) curve analysis was performed to determine the ability of MEA to distinguish between patients with and without the cumulative event in follow-up. The optimal cutoff was calculated by determining the value for platelet aggregation (in arbitrary units) that provided the greatest sum of sensitivity and specificity.
Continuous data are presented as mean and standard deviation and groups were compared with the Student's test. Categorical variables are reported as frequencies and percentages, and the chi-square or Fisher's exact tests were used when appropriate.
Cumulative survival curves were constructed by the Kaplan–Meier method and groups were compared with the log-rank test. The observational period started at hospital discharge.
Multivariate Cox regression analysis was performed for the primary endpoint. Variables that were significant at the bivariate level (with a p value <0.10) and that had clinical relevance were included in the model.
Multivariate logistic regression analysis was performed to identify independent predictors of poor antiplatelet response. Variables with a p value <0.10 on bivariate analysis were included in the model.
All statistical tests were two-tailed and a p value of <0.05 was deemed significant. The analysis was performed with SPSS (Statistical Package for the Social Sciences) version 15, SPSS Inc, Chicago, IL.
ResultsA total of 70 patients were included in the analysis. The median age of the population was 59.9±10.7 years, with a predominance of males (84.3%).
MEA measurements in the study population were normally distributed (Kolmogorov–Smirnov test, p=0.2). The mean ADP-induced platelet aggregation at discharge after an ACS was 17.9±8.6U. A cutoff of 18.5U had the highest sensitivity (75%) and specificity (68%) for the combined endpoint, and the population was therefore divided into two groups based on this value.
The cutoff value for post-treatment MEA measurements defining the upper quartile of patients was 22.3U. According to this value, 17 patients were defined as low responders to clopidogrel.
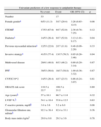
The baseline characteristics of the study population are presented in Table 1. Male gender was more often associated with lower levels of platelet aggregation (90.9 vs. 73.1%, p=0.05). The groups were similar with respect to admission diagnosis, risk factors, previous cardiovascular history, and medical therapy before admission, with the exception of statin therapy. Previous treatment with statins (59.1 vs. 30.8%, p=0.02) was more often associated with a higher response to clopidogrel therapy.
Baseline characteristics of the study population.
| Overall | ADP <18.5 | ADP ≥18.5 | p | |
| Number of patients | 70 | 44 | 26 | |
| Male (%) | 59/70 (84.3) | 40/44 (90.9) | 19/26 (73.1) | 0.05 |
| Age (mean, SD) | 58.8±10.2 | 57.4±10.6 | 60.4±9.6 | 0.21 |
| CYP2C19*2 | 18/70 (25.7) | 11/44 (25.0) | 7/26 (26.9) | 0.86 |
| Admission diagnosis (%) | ||||
| STEMI | 26/70 (37.1) | 13/44 (29.5) | 13/26 (50.0) | 0.09 |
| NSTEMI | 24/70 (34.3) | 18/44 (40.9) | 6/26 (23.1) | 0.13 |
| UA | 20/70 (28.6) | 13/44 (29.5) | 7/26 (26.9) | 0.82 |
| Cardiovascular risk factors (%) | ||||
| Diabetes | 23/70 (32.9) | 11/44 (25.0) | 12/26 (46.2) | 0.07 |
| Dyslipidemia | 44/70 (62.9) | 29/44 (65.9) | 15/26 (57.7) | 0.49 |
| Hypertension | 54/70 (77.1) | 33/44 (75.0) | 21/26 (80.8) | 0.58 |
| Current smoking | 16/70 (22.9) | 9/44 (20.5) | 7/26 (26.9) | 0.53 |
| Cardiovascular history (%) | ||||
| Previous infarction | 14/70 (20.0) | 11/44 (25.0) | 3/26 (11.5) | 0.17 |
| Previous PCI | 10/70 (14.5) | 7/44 (16.3) | 3/26 (11.5) | 0.59 |
| Previous CABG | 3/70 (4.3) | 2/44 (4.5) | 1/26 (3.8) | 0.89 |
| Previous medication (%) | ||||
| Aspirin | 27/70 (38.6) | 16/44 (36.4) | 11/26 (42.3) | 0.62 |
| Other antiplatelets | 11/70 (15.7) | 6/44 (13.6) | 5/26 (19.2) | 0.53 |
| Beta-blockers | 21/70 (0.91) | 13/44 (29.5) | 8/26 (30.8) | 0.91 |
| ACE inhibitors | 35/70 (50.0) | 22/44 (50.0) | 13/26 (50.0) | 1.0 |
| Statins | 34/70 (48.6) | 26/44 (59.1) | 8/26 (30.8) | 0.02 |
| Diuretics | 13/70 (18.6) | 9/44 (20.5) | 4/26 (15.4) | 0.60 |
| Nitrates | 5/70 (7.1) | 4/44 (9.1) | 1/26 (3.8) | 0.41 |
ACE: angiotensin-converting enzyme; CABG: coronary artery bypass grafting; NSTEMI: non-ST elevation acute myocardial infarction; PCI: percutaneous coronary intervention; SD: standard deviation; STEMI: ST-elevation acute myocardial infarction; UA: unstable angina.
The groups were similar regarding hemodynamic data at admission, left ventricular ejection fraction (LVEF), electrocardiographic data, cardiac biomarkers, renal function and lipid profile (Table 2). Platelet and hemoglobin variation at admission were also similar for both subpopulations. High-sensitivity C-reactive protein (hs-CRP) levels were higher in group B (2.6±3.3. vs. 5.1±5.3mg/dl, p=0.02).
Hemodynamic, electrical and laboratory data.
| Overall | ADP <18.5 | ADP ≥18.5 | p | |
| Hemodynamic data on admission | ||||
| Heart rate, bpm* | 74.0±10.2 | 72.9±10.4 | 74.4±10.5 | 0.95 |
| Systolic blood pressure, mmHg* | 137.3±20.6 | 138.2±16.7 | 137.9±26.7 | 0.66 |
| Diastolic blood pressure, mmHg* | 76.4±11.1 | 77.7±10.9 | 76.4±11.5 | 0.56 |
| Body mass index (kg/m2) | 29.2±8.3 | 28.9±5.8 | 31.3±12.3 | 0.28 |
| LVEF (%) | 54.3±10.6 | 54.4±9.4 | 54.6±13.1 | 0.94 |
| Electrical data on admission (%) | ||||
| Sinus rhythm | 68/70 (97.1) | 42/44 (95.5) | 26/26 (100.0) | 0.27 |
| AF | 1/70 (1.4) | 1/44 (2.3) | 0/26 (0.0) | 0.44 |
| ST depression | 16/70 (22.9) | 8/44 (18.2) | 8/26 (30.8) | 0.23 |
| T-wave inversion | 11/70 (15.7) | 7/44 (15.9) | 4/26 (15.4) | 0.95 |
| Laboratory (median, IQ range) | ||||
| Peak troponin I, U/l | 23.9±55.8 | 25.6±66.8 | 14.6±24.4 | 0.42 |
| Peak CKMB mass, U/l | 74.1±166.6 | 77.1±200.5 | 30.8±42.6 | 0.25 |
| Total cholesterol, mg/dl | 196.9±51.6 | 201.1±53.5 | 194.0±52.2 | 0.59 |
| LDL cholesterol, mg/dl | 134.3±39.2 | 136.3±40.0 | 130.3±36.6 | 0.54 |
| Glomerular filtration rate, ml/min | 89.9±36.3 | 93.1±39.5 | 84.1±32.8 | 0.34 |
| Peak C-reactive protein, mg/dl | 3.7±4.4 | 2.6±3.3 | 5.1±5.3 | 0.02 |
| Admission hemoglobin, mg/dl | 13.5±1.7 | 13.4±1.7 | 13.5±1.6 | 0.74 |
| Nadir hemoglobin, mg/dl | 12.4±1.8 | 12.5±1.9 | 12.6±1.6 | 0.80 |
| Hemoglobin variation, % | 7.3±8.1 | 7.5±8.9 | 7.1±6.8 | 0.59 |
| Admission platelets, 109/l | 209.8±62.4 | 199.0±55.9 | 222.8±62.1 | 0.10 |
| Nadir platelets, 109/l | 186.9±58.5 | 175.7±49.2 | 202.1±56.2 | 0.04 |
| Platelet variation, % | 10.4±11.4 | 10.6±12.7 | 8.9±9.1 | 0.59 |
AF: atrial fibrillation; IQ: interquartile; LDL: low-density lipoprotein; LVEF: left ventricular ejection fraction; SD: standard deviation.
GRACE risk scores were similar between subgroups (108.9±23.4 vs. 112.6±20.1, p=0.71). Almost three-quarters of the study population were referred for an invasive strategy, and the subpopulations were also similar regarding rate of invasive strategy, multivessel disease, and use of drug-eluting stents. CYP2C19*2 allele frequency was also similar in both patient groups (Table 3).
Risk score assessment and in-hospital management.
| Overall | ADP <18.5 | ADP ≥18.5 | p | |
| Length of hospital stay, days | 4.4±1.5 | 4.4±1.4 | 4.7±1.7 | 0.56 |
| GRACE risk score | 110.8±24.3 | 108.9±23.4 | 112.6±20.1 | 0.71 |
| TIMI score ≤2 | 44/70 (62.9) | 29/44 (65.9) | 15/26 (57.7) | 0.49 |
| TIMI score 3 or 4 | 21/70 (30.0) | 13/44 (29.5) | 8/26 (30.8) | 0.91 |
| TIMI score ≥5 | 5/70 (7.1) | 2/44 (4.5) | 3/26 (11.5) | 0.27 |
| Invasive strategy (%) | 54/70 (77.1) | 34/44 (77.3) | 20/26 (76.9) | 0.97 |
| Non-significant coronary disease (%) | 6/54 (11.1) | 4/34 (11.8) | 2/20 (10.0) | 0.84 |
| One-vessel disease (%) | 22/54 (40.7) | 14/34 (41.2) | 8/20 (40.0) | 0.93 |
| Two-vessel disease (%) | 11/54 (20.4) | 7/34 (20.6) | 4/20 (20.0) | 0.96 |
| Three-vessel disease (%) | 15/54 (27.8) | 9/34 (26.5) | 6/20 (30.0) | 0.78 |
| Stent (%) | 38/54 (70.4) | 25/34 (73.5) | 13/20 (65.0) | 0.51 |
| Drug-eluting stent (%) | 29/38 (76.3) | 21/25 (84.0) | 8/13 (61.5) | 0.12 |
The groups were also similar in terms of medical treatment during hospital stay and at discharge. Low molecular weight heparin was used in the majority of patients, and GP IIb/IIIa inhibitors were prescribed in a third of the overall population. There was a very high rate of prescription of statins, beta-blockers and ACE inhibitors at discharge (Table 4).
Medical therapy.
| Overall | ADP <18.5 | ADP ≥18.5 | p | |
| Antiplatelet and antithrombotic medication in the first 24hours (%) | ||||
| Aspirin | 68/70 (97.1) | 43/44 (97.7) | 25/26 (96.2) | 0.71 |
| Clopidogrel | 69/70 (98.6) | 43/44 (97.7) | 26/26 (100.0) | 0.44 |
| LMWH | 67/70 (95.7) | 43/44 (97.7) | 24/26 (92.3) | 0.28 |
| Fondaparinux | 6/70 (7.7) | 4/44 (9.1) | 2/26 (7.7) | 0.84 |
| GP IIb/IIIa inhibitors | 23/70 (32.9) | 15/44 (34.1) | 8/26 (30.8) | 0.78 |
| Anti-ischemic medication in the first 24hours (%) | ||||
| Beta-blockers | 67/70 (95.7) | 43/44 (97.7) | 24/26 (92.3) | 0.28 |
| Nitrates | 9/70 (12.9) | 7/44 (15.9) | 2/26 (7.7) | 0.32 |
| Calcium channel blockers | 8/70 (11.4) | 7/44 (15.9) | 1/26 (3.8) | 0.13 |
| Other therapy in the first 24hours (%) | ||||
| ACE inhibitors/ARBs | 67/70 (95.7) | 43/44 (97.7) | 24/26 (92.3) | 0.28 |
| Statins | 70/70 (100.0) | 44/44 (100.0) | 26/26 (100.0) | |
| Diuretics | 5/70 (7.1) | 4/44 (9.1) | 1/26 (3.8) | 0.41 |
| Proton pump inhibitors | 40/70 (57.1) | 28/44 (63.6) | 12/26 (46.2) | 0.15 |
| Medical therapy at discharge (%) | ||||
| Aspirin | 70/70 (100.0) | 44/44 (100.0) | 26/26 (100.0) | |
| Clopidogrel | 70/70 (100.0) | 44/44 (100.0) | 26/26 (100.0) | |
| Beta-blockers | 64/70 (91.4) | 39/44 (88.6) | 25/26 (96.2) | 0.28 |
| ACE inhibitors/ARBs | 66/70 (94.3) | 42/44 (95.5) | 24/26 (92.3) | 0.58 |
| Statins | 66/70 (94.3) | 42/44 (95.5) | 24/26 (92.3) | 0.58 |
ACE: angiotensin-converting enzyme; ARBs: angiotensin receptor blockers; GP: glycoprotein; LMWH: low molecular weight heparin.
During follow-up there were eight primary endpoints. One patient died, four had new non-fatal myocardial infarction, and three were readmitted for unstable angina.
Cumulative freedom from the combined result was 96.0% for group A, vs. 76.7% for group B (log-rank p<0.01) (Figure 1). We identified two independent predictors of the primary result: a poor clopidogrel metabolizer genotype (HR 3.97, 95% CI 1.05–15.04, p=0.04), and ADP-induced platelet aggregation measured by MEA at discharge ≥18.5U (HR 6.75, 95% CI 1.38–32.90, p=0.02), in a model adjusted for LVEF, renal function, and diabetes (Table 5).

Univariate and multivariate predictors of the combined event.
| Univariate predictors of the combined event: | ||||
| No event | Event | OR (95% CI) | p | |
| Number | 59 | 8 | ||
| Female gendera | 8/59 (13.6) | 3/8 (37.5) | 3.83 (0.76–19.20) | 0.11 |
| STEMIa | 23/59 (39.0) | 2/8 (25.0) | 0.52 (0.10–2.81) | 0.44 |
| Diabetesa | 18/59 (30.5) | 5/8 (62.5) | 3.80 (0.82–17.6) | 0.07 |
| Previous myocardial infarctiona | 12/59 (20.3) | 2/8 (25.0) | 1.31 (0.23–7.30) | 0.76 |
| Invasive strategya | 45/59 (76.3) | 7/8 (87.5) | 2.18 (0.25–19.23) | 0.48 |
| Multivessel diseasea | 22/45 (48.9) | 4/7 (57.1) | 1.39 (0.28–6.95) | 0.69 |
| Drug-eluting stentsa | 24/32 (75.0) | 3/4 (75%) | 1.00 (0.55–1.82) | 1.00 |
| PPIa | 35/59 (59.3) | 3/8 (37.5) | 0.41 (0.10–1.90) | 0.24 |
| CYP2C19*2 | 11/59 (18.6) | 5/8 (62.5) | 7.27 (1.51–35.0) | 0.01 |
| ADP-induced platelet aggregation ≥18.5 | 19/59 (32.2) | 6/8 (75.0) | 6.32 (1.16–34.3) | 0.02 |
| Age (years)b | 60.2±9.5 | 60.3±10.9 | 0.98 | |
| LVEF %,b | 60.4±14.2 | 67.1±15.3 | 0.19 | |
| Glomerular filtration rate, ml/minb | 90.0±35.8 | 79.5±36.1 | 0.39 | |
| Peak troponin I, U/L | 22.9±52.4 | 11.8±17.3 | 0.51 | |
| Multivariate Cox regression analysis for the composite endpoint at follow-up | |||
| Variables | HR | p | 95% CI |
| Diabetes | 0.96 | 0.96 | 0.24–3.95 |
| CYP2C19*2 | 3.97 | 0.04 | 1.05–15.04 |
| ADP ≥18.5U | 6.75 | 0.02 | 1.38–32.90 |
| Chi-square 12.6; p<0.01 | |||
LVEF: left ventricular ejection fraction; PPI: proton pump inhibitors; STEMI: ST-elevation acute myocardial infarction.
Multivariate logistic regression analysis was performed to identify independent predictors of low response to clopidogrel (Table 6). The model included platelet response to clopidogrel, but also potential confounding variables in the patients’ baseline characteristics. The model only identified one independent predictor of poor response to clopidogrel, admission hs-CRP of over 4.3mg/dl (adjusted OR 3.43, 95% CI 1.06–14.61, p=0.04).
Univariate and multivariate predictors of a low response to antiplatelet therapy.
| Univariate predictors of a low response to antiplatelet therapy | ||||
| No event | Event | OR (95% CI) | p | |
| Number | 53 | 17 | ||
| Female gendera | 6/53 (11.3) | 5/17 (29.4) | 3.26 (0.85–12.5) | 0.08 |
| STEMIa | 17/53 (67.9) | 9/17 (52.9) | 2.38 (0.78–7.25) | 0.12 |
| Diabetesa | 14/53 (26.4) | 9/17 (52.9) | 3.13 (1.01–9.17) | 0.04 |
| Previous myocardial infarctiona | 12/53 (22.6) | 2/17 (11.8) | 0.46 (0.09–2.28) | 0.33 |
| Invasive strategya | 41/53 (77.4) | 13/17 (76.5) | 0.95 (0.26–3.46) | 0.94 |
| Multivessel disease | 20/41 (48.8) | 6/13 (46.2) | 0.90 (0.29–3.14) | 0.87 |
| PPIa | 30/53 (56.6) | 10/17 (58.8) | 1.09 (0.36–3.32) | 0.87 |
| CYP2C19*2 | 14/53 (26.4) | 4/17 (23.5) | 0.86 (0.24–3.07) | 0.81 |
| GRACE risk score | 110.5±22.7 | 108.5±21.2 | 0.80 | |
| Age (years)b | 57±10.1 | 60.7±11.0 | 0.32 | |
| LVEF %,b | 54.1±10.4 | 55.6±12.5 | 0.63 | |
| C-reactive protein, mg/dlb | 3.0±3.6 | 5.2±6.0 | 0.08 | |
| Glomerular filtration rate, ml/minb | 92.3±38.4 | 82.4±33.1 | 0.36 | |
| Body mass index kg/m2 | 29.9±9.8 | 29.3±3.6 | 0.78 | |
| Multivariate logistic regression: predictors of a low response to antiplatelet therapy | |||
| Variables | OR | p | 95% C I |
| CRP ≥4.3mg/dl | 3.93 | 0.04 | 1.06–14.61 |
| Female gender | 4.40 | 0.06 | 0.93–20.72 |
| Diabetes | 2.19 | 0.21 | 0.65–7.38 |
| C-statistic: 0.72; Hosmer–Lemeshow: p=0.85 | |||
CRP: C-reactive protein; LVEF: left ventricular ejection fraction; PPI: proton pump inhibitors; STEMI: ST-elevation acute myocardial infarction.
Our study showed that in a single-center ACS population, a platelet aggregation level ≥18.5U at discharge assessed by MEA on a Multiplate analyzer was an independent predictor of a combined ischemic endpoint. Importantly, this analysis also confirmed the role of inflammation in influencing platelet response to clopidogrel.
Different methods are currently available to assess platelet function. LTA and the VerifyNow assay are based on light transmission in a liquid phase after ADP stimulation, whereas MEA works on the principles of platelet aggregometry.18 As previously reported, there is a good correlation between LTA and MEA values10 and between VerifyNow and MEA.19
Assessing platelet function in whole blood (MEA and VerifyNow) has some advantages over LTA, which requires platelet-rich plasma. There is no need for centrifugation, which can alter platelet function, to separate platelets from other blood cells. In contrast to LTA or VerifyNow, in which aggregation occurs in a liquid phase, aggregation in MEA takes place on surfaces. This is similar to in vivo conditions, in which platelet aggregation also takes place on surfaces, such as on ruptured plaques, at sites of vascular injury, or on stent struts.5,20
The optimal method to quantify platelet reactivity as well as the definition of the threshold for high on-treatment platelet reactivity to ADP has been the subject of controversy. According to previous studies, any definition of high on-treatment platelet reactivity will only be meaningful when a cutoff or target value is identified by an accepted statistical test.21 Generally, ROC curve analysis has been used to define the optimal cutoff to define high on-treatment platelet reactivity associated with ischemic risk, providing the greatest sum of sensitivity and specificity, including prospective studies of individualized antiplatelet therapy in PCI patients.22 We therefore assessed platelet function at discharge, and used a ROC curve to define the best cutoff for the follow-up ischemic endpoint.
Prognostic ischemic relevanceIn our patient cohort two independent predictors of medium-term ischemic outcome were identified: platelet aggregation ≥18.5U and poor clopidogrel metabolizer genotype.
It is difficult to compare our data with previous studies, as many variables differ, including populations, the timing of the clopidogrel reactivity assessment, the method used and the endpoint analyzed.21
Nevertheless, the area under the curve (AUC) of MEA measurement of clopidogrel resistance in our study (0.75, 95% CI 0.62–0.87, p=0.01) was similar to that reported by Sibbing et al. (AUC of 0.78) in their paper on the importance of MEA in predicting stent thrombosis 30 days after PCI,5 with higher sensitivity (75%–70%) but lower specificity (68%–84%). Also, comparing different platelet function tests,23 the AUC gave consistent results for platelet function tests and ischemic outcome.
As we have previously reported, age is one of the most important variables when considering risk assessment in follow-up analysis.24 We decided to exclude patients over 75 years of age, and considered only those surviving hospital stay. Our population consequently had a lower mean age than previous studies, higher LVEF, and a lower overall risk assessment (by TIMI and GRACE scores), thereby highlighting the role of platelet function in medium-term follow-up.
Definition of non-response to clopidogrelMeasurement of clopidogrel responsiveness (absolute or relative changes in platelet aggregation from baseline) appears to be the most reliable indicator of a treatment effect, but it may not optimally identify patients at risk.21 Given the interindividual variability in baseline ADP-induced platelet aggregation, measurement of clopidogrel responsiveness could overestimate ischemic risk in nonresponders with low pretreatment reactivity, as well as underestimating risk in responders who still have high platelet reactivity after treatment.25 Therefore, the absolute level of platelet reactivity during treatment (on-treatment platelet reactivity) has been proposed as a better measure of thrombotic risk than responsiveness to clopidogrel.21 This hypothesis influenced our decision to assess platelet reactivity at discharge following an ACS.
The definition of low response to clopidogrel varies from study to study, and most previous authors have used the upper 5–40% of patients to define a cutoff for low response. Previous studies using MEA measurement with the Multiplate analyzer have defined low clopidogrel response by setting a cutoff at the upper quintile of MEA measurements.5 Given the small size of our study population, we defined a nonresponder to clopidogrel therapy as one within the highest quartile of the MEA distribution.
We found that an inflammatory state as assessed by CRP level was an independent predictor of a low response to clopidogrel. ACS are known to result from destabilization, disruption, or rupture of an atherosclerotic plaque.26 Inflammation is believed to play a decisive initial and perpetuating role in the thrombotic transformation of this plaque rupture, and platelet activation is central to the formation of thrombus. This increased state of inflammation is paralleled by activation of platelets with surface-exposed P-selectin, which tethers platelets to monocytes via P-selectin glycoprotein ligand-1 to form platelet–monocyte aggregates. The close interaction between monocytes and platelets leads to leukocyte activation and expression of cell adhesion molecules, and activates the release of enzymes such as matrix metalloproteinases which degrade the subendothelial basement membrane, leading to plaque rupture or thrombogenicity.27
Interestingly, our data associated previous statin therapy with a better response to clopidogrel. While LDL cholesterol reduction has been considered the primary goal of lipid-lowering therapy, the pleiotropic effects of statins including plaque stabilization, reversal of endothelial dysfunction, and reduced inflammation (by decreasing CRP) may confer additional benefits.28 Moreover, statins appear to have important effects in thrombogenesis, reducing expression of tissue factor production and activity, increase production of tissue factor package inhibitor, decrease platelet thrombus formation and improve fibrinolysis as a result of lower plasminogen activator inhibitor-1 levels.29
Our sample was small and this was probably the reason that a poor metabolizer genotype was not a predictor of a poor platelet response to clopidogrel. By contrast, our data revealed the CYP2C19*2 allele to be an independent predictor of medium-term outcome. This was in agreement with previous data that reported prognostic importance for carriers of at least one reduced-function CYP2C19 allele, whether in the context of ACS or of clopidogrel-treated patients with stable coronary artery disease.30
Future directionsPrevious trials have proved the clinical benefit of achieving lower levels of on-treatment platelet reactivity, as suggested by the TRITON–TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel–Thrombolysis In Myocardial Infarction 38)31 and the PLATO (Platelet Inhibition and Patient Outcomes) trials.32 This was also corroborated by a 2011 meta-analysis that included over 3000 patients.33
The recently presented randomized controlled trial, GRAVITAS (Gauging Responsiveness with A VerifyNow assay--Impact on Thrombosis And Safety), which used the VerifyNow platelet assay in a population with stable coronary disease after PCI, concluded that there were no benefits in terms of cardiovascular outcomes or stent thrombosis with a double dose of clopidogrel in patients receiving drug-eluting stents with high residual platelet activity compared to the regular clopidogrel dose.34 The recommendations published in 2009 by the European Society of Cardiology thus remain valid: “… there are no clinical data obtained from prospective trials in sufficiently large number of patients, showing that the routine or even the occasional determination/monitoring of platelet function while on therapy with antiplatelet drugs and consequent therapeutic decisions leads to any practical clinically relevant advantage”.35 In other words, the best treatment strategy for high platelet reactivity remains unknown.
LimitationsPlatelet function tests were not performed prior to clopidogrel therapy. Patients with non-ST elevation ACS received 300mg of clopidogrel, and those with ST-elevation ACS received a loading dose of 600mg. Although MEA measurements correlate with those using LTA and VerifyNow, it is not possible to extrapolate our results to other assays. The small size of our population made it impossible to determine the relevance of MEA measurements regarding bleeding complications or other endpoints such as stent thrombosis.
ConclusionIn our ACS study population a low response to clopidogrel assessed by the Multiplate analyzer was an independent predictor of a medium-term ischemic outcome after an ACS. Inflammation was an independent predictor of a lower clopidogrel response.
Conflicts of interestThe authors have no conflicts of interest to declare.
The authors would like to thank Mrs Alda Reboredo for the logistic support of the study.
