
Takotsubo cardiomyopathy is a reversible condition, characterized by transient left ventricular systolic dysfunction, that mimics an acute coronary syndrome. It usually occurs after physical or emotional stress, predominantly in postmenopausal women, although it also can affect younger age groups and males. It often presents as chest pain or dyspnea with electrocardiographic changes and mild elevation of cardiac enzymes suggesting acute myocardial infarction. Coronary angiography excludes obstructive coronary disease, and imaging reveals ventricular apical akinesia and compensatory hypercontractility of the basal segments.
Various pathophysiological mechanisms have been proposed for the syndrome, such as occult atherosclerotic disease, multivessel spasm and/or microvascular dysfunction. However, the most widely accepted hypothesis at present is an excess of catecholamines causing calcium overload in cardiac myocytes, leading to disruption of contraction and ventricular function. Treatment is essentially supportive, with spontaneous and complete reversal of the changes within days or weeks. However, the presence of complications and comorbidities may predict a more adverse prognosis. As much is still unknown about takotsubo cardiomyopathy and the number of reported cases is growing, we present a literature review.
A miocardiopatia Takotsubo é uma situação reversível, caracterizada por disfunção sistólica transitória do ventrículo esquerdo, a qual mimetiza uma síndrome coronária aguda. Ocorre habitualmente após stress físico ou emocional, com predomínio em mulheres na pós-menopausa, apesar de também poder afetar faixas etárias mais jovens e o género masculino. Apresenta-se frequentemente com dor torácica ou dispneia, alterações eletrocardiográficas sugestivas de enfarte agudo do miocárdio e ligeira elevação das enzimas cardíacas. A angiografia coronária exclui doença arterial obstrutiva, constatando-se, imagiologicamente, acinesia apical ventricular, com hipercontratilidade basal «compensatória».
Foram propostos vários mecanismos fisiopatológicos para a síndrome, como doença aterosclerótica oculta, espasmo multivasos ou disfunção microvascular. Contudo, a hipótese mais aceite atualmente é a existência de um excesso de catecolaminas que, por causar uma sobrecarga de cálcio nos miócitos cardíacos, leva a perturbação da contração e função ventriculares. O tratamento é essencialmente de suporte, com reversão espontânea e completa das alterações num intervalo de dias a semanas. Contudo, a ocorrência de complicações e algumas comorbilidades podem predizer um prognóstico menos benigno. Assim, dado que a síndrome Takotsubo ainda é desconhecida em muitos aspetos e face ao crescente número de casos descritos, fez-se uma revisão da literatura do conhecimento atual.
For more than three decades, heart muscle diseases were classified into primary or idiopathic myocardial diseases (cardiomyopathies) and secondary or specific diseases (those of known etiology or associated with systemic disorders).1,2 This classification was revised by the American Heart Association,2 but the European Society of Cardiology subsequently proposed what it believes is a clinically more useful classification system, based on ventricular morphology and function.1,2 This defines a cardiomyopathy as a myocardial disorder in which the heart muscle is structurally and functionally abnormal, in the absence of coronary artery disease (CAD – Table 1), hypertension, valvular disease and congenital heart disease sufficient to cause the observed abnormality. Cardiomyopathies are grouped into specific morphological and functional phenotypes – hypertrophic, dilated, arrhythmogenic right ventricular, restrictive and unclassified – and each phenotype is then sub-classified into familial and non-familial forms.1
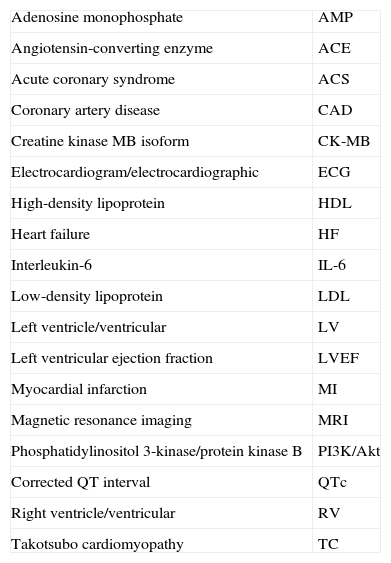
List of abbreviations.
| Adenosine monophosphate | AMP |
| Angiotensin-converting enzyme | ACE |
| Acute coronary syndrome | ACS |
| Coronary artery disease | CAD |
| Creatine kinase MB isoform | CK-MB |
| Electrocardiogram/electrocardiographic | ECG |
| High-density lipoprotein | HDL |
| Heart failure | HF |
| Interleukin-6 | IL-6 |
| Low-density lipoprotein | LDL |
| Left ventricle/ventricular | LV |
| Left ventricular ejection fraction | LVEF |
| Myocardial infarction | MI |
| Magnetic resonance imaging | MRI |
| Phosphatidylinositol 3-kinase/protein kinase B | PI3K/Akt |
| Corrected QT interval | QTc |
| Right ventricle/ventricular | RV |
| Takotsubo cardiomyopathy | TC |
Takotsubo cardiomyopathy (TC) is an unclassified, non-familial form.1 Originally described in Japan, it is considered to be stress-related; it is characterized by acute onset of symptoms and electrocardiographic (ECG) alterations that mimic myocardial infarction (MI), and mild elevation of myocardial enzymes, usually following physical or emotional stress.3,4 The transient left ventricular (LV) dysfunction seen in these patients, with apical and medial akinesia and basal hyperkinesia, gives the LV the characteristic shape of a takotsubo, a pot with a round bottom and narrow neck used for trapping octopuses in Japan.5 Most patients recover completely in days or weeks.4
Various pathophysiological mechanisms have been proposed for this entity, but much is still unknown about these mechanisms as well as about treatment and prognosis; there is also uncertainty concerning the differential diagnosis with MI. Given these questions and the growing number of reported cases, we present a literature review.
MethodologyWe searched the PubMed database for articles published in English between 2005 and 2011 using the search terms takotsubo cardiomyopathy and takotsubo cardiomyopathy prognosis (with no additional limits). For the first terms 1020 results were obtained, of which the 200 most recent articles were selected, and 150 were obtained for the second terms. To be included in the analysis each article had to be about takotsubo cardiomyopathy (even if referred to by another name) and contain data relevant to at least one of the following: prevalence, clinical presentation, natural history, prognosis, and underlying pathophysiological mechanisms.
The titles and/or abstracts of the publications were reviewed and those that appeared to meet the conditions and objectives of the study were analyzed in detail. Further suitable articles were added following searches of the New England Journal of Medicine database and the websites of the European Society of Cardiology and the Portuguese Society of Cardiology. The final list for review consisted of 40 articles. Given the heterogeneity of their methods and results, it was decided to perform a descriptive analysis.
PresentationTakotsubo cardiomyopathy, first described in 1991 in the Japanese population, is a reversible condition that is often precipitated by a stressful event.7 It was initially named after the Japanese octopus pot because of the similarity in shape of the LV during systole due to wall motion abnormalities,5 but as the number of cases around the world has grown, various other terms have been proposed, including apical ballooning syndrome, transient left ventricular dysfunction with apical ballooning and broken heart syndrome, among a total of 75 different names. However, the original name seems most appropriate, since it is general enough to allow future clinical variants to be included within this nomenclature, recalls the characteristic alterations in LV morphology, and acknowledges the Japanese investigators who first described it.6
The clinical picture of TC is frequently indistinguishable from that of an acute coronary syndrome (ACS).5 The most common presenting symptom is acute onset of chest pain, which is sometimes accompanied by dyspnea, palpitations, diaphoresis, nausea, vomiting or syncope.5,7 Less frequently, there may be arrhythmias or hemodynamic instability with hypotension or cardiogenic shock, requiring circulatory and ventilatory assistance.4,5,8
These symptoms are often preceded by emotional stress (assault, financial loss, unexpected death of a relative or friend, confrontational arguments or divorce) – hence the term “broken heart syndrome” – or physical stress such as pulmonary disease, sepsis, trauma, postoperative state or hyperthyroidism.5,9–11 A case of TC was recently reported that was triggered by watching a 3D action film, the first published association between the syndrome and visual stimulation.10 Catecholamine excess due to pheochromocytoma can also cause the morphological and functional changes of TC.12 However, in a third of cases no preceding trigger is found.5,7
TC mainly affects postmenopausal women (around 90% of cases), and the average age of those affected is 62–76 years.5–7 However, a higher proportion of premenopausal women are affected by the apical-sparing variant of the disease, while a male predominance has been reported in cases precipitated by physical stress.5
The real prevalence of TC is unknown, but it is estimated to account for 1–2% of cases presenting as ACS. It has been reported in North and South America, Europe, Asia, South Africa and Australia.4,5,7
Electrocardiographic features and cardiac biomarkersECG findings usually mimic those of MI; ST-segment elevation has been reported in 56%, T-wave inversion in 17% and Q-waves or abnormal R-wave progression in 10%; the remaining 17% have either non-specific changes or no changes at all.5 The most frequent alterations as the condition evolves are new or deepening T-wave inversion, most prominently in leads V2–V6 (in the first 2–3 days and persisting for 2–3 weeks) and prolonged corrected QT interval (QTc).5,7 These changes usually disappear completely within three months.13
The absence of reciprocal changes in the inferior leads and a ratio of ≥1 between ST-segment elevation in leads V4–V6 and V1–V3 has been considered highly specific for TC. Other authors have reported that the magnitude of the ST elevation is less in TC patients, and that they develop deeper T-wave inversion or more prolonged QTc than CAD patients. These findings, however, are too subtle to be helpful in the differential diagnosis between TC and ACS.5,7
Cardiac enzymes (troponin I and T and CK-MB) are slightly elevated in most cases, although the absence of enzyme elevation does not exclude the diagnosis. This finding is suggestive of myocardial injury, which in this context would call for coronary angiography. Troponin T levels peak in the first 24h, followed by a gradual fall5; B-type natriuretic peptide, which reflects LV systolic dysfunction, is also elevated in TC, with higher levels than in ST-elevation MI, and this, together with the relatively modest rise in troponin T and the large area of LV dysfunction, can help distinguish TC from MI.7,8
Imaging featuresCoronary angiography typically shows normal coronary arteries or non-obstructive CAD (luminal stenosis <50%),4,7 but should be performed to exclude thrombus or acute plaque rupture and is thus valuable in differential diagnosis with MI. In a small percentage of patients TC can coexist with obstructive coronary disease, a reflection of the high prevalence of CAD in elderly populations.7
Echocardiography and ventriculography help clarify the situation and confirm LV systolic dysfunction, and in the classic form of TC reveal extensive apical and/or midventricular akinesia or hypokinesia but with basal function preserved or hyperkinetic, resulting in the characteristic appearance (narrow base with apical ballooning) that gives the condition its name.4,5 These wall motion abnormalities typically extend beyond the vascular territory of a single coronary artery, suggesting that myocardial stunning rather than necrosis is the underlying mechanism of the acute LV dysfunction. Left ventricular ejection fraction (LVEF) is also diminished at admission, with values ranging from 20 to 49%.4,7 A transient dynamic intraventricular pressure gradient is sometimes found.4 Repeat assessment 6–8 weeks later shows complete recovery of LV function in terms of wall motion and LVEF.7
Cardiac magnetic resonance imaging (MRI) can be particularly helpful, since it can demonstrate the absence of myocardial necrosis when there is no late gadolinium enhancement; it is also the best tool for diagnosing right ventricular (RV) involvement in TC, which was once considered rare but is reported with increasing frequency (26–30% of patients).5,14 RV systolic dysfunction is associated with lower LVEF, more frequent apical involvement and pleural effusion; the variable involvement of this ventricle has been attributed to differences in the intensity of the initial trigger or in blood supply.7,14
DiagnosisIn 2003, Abe and Kondo proposed diagnostic criteria for TC, divided into major (LV wall motion and ECG abnormalities), minor (stress as triggering factor, elevation of cardiac enzymes and chest pain) and exclusion (including subarachnoid hemorrhage, pheochromocytoma and myocarditis).8
In 2004, guidelines were approved for the diagnosis of TC, developed by the Japanese Idiopathic Cardiomyopathy Research Committee and based on questionnaires sent to researchers in the field. They included a definition of TC, exclusion criteria and references for diagnosis.15
However, diagnostic criteria are being updated with advances in understanding of TC. For example, since TC can occur in younger women and in men and with no stress trigger, age, gender and the presence of a triggering factor have not been included in more recent criteria; nor has documented reversibility of LV systolic dysfunction, since the intention is to establish the diagnosis at hospital admission.7
A group of specialists at the Mayo Clinic in Rochester, MN, USA, have accordingly proposed a set of four diagnostic criteria that must be present at admission for a diagnosis of TC, and which are now most often used as a reference in published studies:
- •
Transient hypokinesia, akinesia, or dyskinesia of the left ventricular mid segments with or without apical involvement; the regional wall motion abnormalities extend beyond a single epicardial vascular distribution; a stressful trigger is often, but not always present;
- •
Absence of obstructive coronary disease or angiographic evidence of acute plaque rupture;
- •
New electrocardiographic abnormalities (either ST-segment elevation and/or T-wave inversion) or modest elevation in cardiac troponin;
- •
Absence of pheochromocytoma or myocarditis.7
The pathophysiological mechanisms underlying TC have been the subject of many studies and several theories have been proposed. One attributes the syndrome to the presence of occult coronary atherosclerosis and plaque rupture leading to acute ischemia; this has been advocated by Ibanez et al., who reported the presence of a long left anterior descending coronary artery, wrapped around the apex, which could explain the extensive apical akinesia seen in TC.5 However, the prevalence of this anatomical variant is low in TC patients, and the development of even transient myocardial ischemia, especially in the setting of occult CAD, requires the presence of an intracoronary thrombus, a feature that has never been demonstrated in these patients.5,7
Another theory is that of coronary spasm, which would have to occur in multiple vessels in order to explain the wall motion abnormalities extending beyond the territory of a single epicardial coronary artery. The evidence for this hypothesis is contradictory; some investigators have succeeded in provoking multivessel spasm in TC patients with infusion of ergonovine or acetylcholine, while others have failed.4,5
Coronary microvascular dysfunction has also been thought to be the cause of TC, on the basis of studies showing abnormal myocardial perfusion and diminished coronary flow in these patients.5,13,16 Severely reduced apical fluorodeoxyglucose uptake on positron emission tomography has also been reported, suggestive of a metabolic defect that could have more impact than perfusion abnormalities. At all events, it remains unknown whether microvascular dysfunction plays a causative role in TC or whether it is a consequence of a primary myocardial impairment process.5
The most widely accepted theory at present is increased local release of catecholamines, leading to adrenergic stimulation of the myocardium and hence wall motion abnormalities.3
Emotional and physical stress can induce excitation of the limbic system. The amygdala and hippocampus, which together with the insula are the principal brain areas involved in emotion and memory, play a central role in the control of cardiovascular function. Their excitation stimulates the medullary autonomic centers, which in turn excite pre- and post-synaptic neurons leading to the release of norepinephrine and its neuronal metabolites. Epinephrine release is simultaneously induced from the adrenal medulla. These catecholamines stimulate the heart through both cardiac and extracardiac sympathetic nerves and via the bloodstream, bind to vessel adrenoreceptors and induce catecholamine toxicity in cardiomyocytes.3
This hypothesis was put forward by Wittstein et al.,17 who observed a significant increase in plasma epinephrine, norepinephrine, dihydroxyphenylglycol and dihydroxyphenylacetic acid in TC patients, consistent with enhanced catecholamine synthesis, neuronal reuptake, and neuronal metabolism. A significant increase in neuropeptide Y, which is stored in postganglionic sympathetic nerves, was also found. By contrast the increase in plasma levels of metanephrine and normetanephrine, which are extraneuronal catecholamine metabolites, was within the same range as that observed in Killip class III myocardial infarction patients, suggesting that cardiac toxicity is mediated more by catecholamines released directly into the heart via neural connections than by those reaching the heart via the bloodstream.3
Sex hormones may exert important influences on the sympathetic neurohormonal axis and on coronary vasoreactivity, since older women appear to be more vulnerable to sympathetically mediated myocardial stunning, and post-menopausal alteration of endothelial function in response to reduced estrogen levels has been suggested as a possible explanation.4 Recent animal models suggest that estrogen suppresses myocardial adrenergic receptors, attenuates the hypothalamo-sympathoadrenal axis, and promotes the production of cardioprotective substances.18
Catecholamines can decrease the viability of cardiomyocytes through cyclic AMP-mediated calcium overload and oxygen-derived free radicals. This hypothesis is supported by the myocardial histological findings of contraction band necrosis, neutrophil infiltration, and fibrosis, which would explain the ventricular dysfunction.3 The typical pattern of apical akinesia with preserved basal contractile function could be explained by the fact that while the base has a greater density of sympathetic nerves and higher norepinephrine content, the apex may be more vulnerable to sympathetic stimulation, possibly because of its higher density of beta-adrenoreceptors.4,5
These apical and mid-ventricular wall motion abnormalities, with hyperkinesia or normal motion of the basal segments, can induce transient dynamic obstruction of the LV outflow tract.3 This obstruction could be seen as a cause rather than a consequence of TC: in a setting of dehydration or massive catecholamine surge due to physical or emotional stress, elderly women, who frequently have a sigmoid septum, could develop severe obstruction, leading to apical ischemia as a result of increased wall stress and reduced subendocardial coronary flow.5,19 There is, however, no progression to subendocardial infarction because as the ischemia becomes more severe, contractile function will be so impaired that the force exerted by the myocardium on the walls of the high-pressure chamber will diminish, reducing the intracavitary gradient and allowing coronary flow to resume.19 Nevertheless, such obstruction is not observed in all cases of TC, and this mechanism cannot explain the apical sparing variants of the disease.5
Reported morphological variants of the LV in TC include isolated midventricular dysfunction with apical and basal sparing, known as midventricular ballooning syndrome, and isolated basal hypokinesia, also known as inverse TC.3,5,20,21 It is not known what causes these different distributions of wall motion abnormalities, but it may be related to differences in autonomic innervation and adrenergic stimulation in the heart or in interindividual variations in the location and density of receptors.3,22 These variants can occur in the same individual, recurrences with different patterns being attributed to changes in receptor sensitivity or stress and catecholamine levels in different situations.22
TC appears to be more frequent during the morning and in the summer, which has been attributed to the relationship between catecholamine levels and chronobiological patterns, which may be useful for prevention at these particularly vulnerable times.23
Subarachnoid hemorrhage and pheochromocytoma can also cause reversible LV wall motion abnormalities that resemble TC, presumably through local release of norepinephrine in the heart. It is thus important to exclude these conditions in cases of suspected TC.12,13
Although myocarditis has been put forward as a cause of TC, this has been ruled out by the fact that neither endomyocardial biopsy nor cardiac MRI has revealed findings compatible with this hypothesis, and viral serology has been negative in all patients tested.4,7
There is thus no single, clear and unequivocal definition of the pathophysiology of TC; information on the syndrome is limited and based mainly on case reports and case series, frequently comprising few patients, and most studies are retrospective with short follow-up periods after hospital discharge.4
Cellular and molecular aspectsGiven the similarities in clinical presentation of TC and MI, various studies have sought to investigate possible differences at the cellular and molecular levels that could help to distinguish the two conditions and to clarify the pathophysiology of TC.
Pirzer et al.24 showed that concentrations of inflammatory mediators and markers of platelet and monocyte activity can help differentiate between the two entities. The platelet-activation marker CD62P, which is directly involved in platelet binding to endothelial cells and leukocytes, is reduced in TC compared to MI, as is admission interleukin-6 (IL-6), a key inflammatory factor in the pathogenesis of atherosclerosis and hence of CAD. This suggests that inflammatory and thrombotic responses play a minor role in TC.
Further evidence that the etiology of TC does not involve atherosclerotic occlusion of the coronary arteries comes from studies showing significantly higher levels of HDL cholesterol and lower LDL cholesterol and triglycerides in patients with TC compared to those with MI, which may also have therapeutic implications in the sense that lipid-lowering therapy should be individualized on a case-by-case basis.25
TC is accompanied by severe morphological changes potentially resulting from catecholamine excess followed by microvascular dysfunction and direct cardiotoxicity. However, these alterations, which occur in proteins such as α-actinin, actin and titin that are important in contractile function, reverse within days, with almost complete normalization of intracellular structure and recovery of ventricular function.26
This rapid recovery of contractile function is demonstrated by studies showing activation of the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) signaling pathway in TC. This activated cell survival cascade protects cardiomyocytes from cell death and may also contribute to the rapid regeneration seen in TC, by directly inhibiting apoptosis and proapoptotic transcription factors and stimulating antiapoptotic factors and cell metabolism. However, it is not known whether this activity is favorable: increased cell survival, which is beneficial for tissue repair, may also lead to unregulated cell growth and tumorigenesis, and further investigation is thus required.27
TreatmentThere is currently no standard treatment for TC. In the acute phase therapy should be directed at resolving myocardial ischemia. It is extremely important to perform cardiac catheterization immediately, since the diagnosis of TC requires the exclusion of obstructive CAD in order to avoid unnecessary thrombolysis. Furthermore, given the similar presentation of TC and MI and the impossibility of distinguishing between them at admission, patients should not be denied the benefits of primary angioplasty. For the same reason, aspirin, clopidogrel, nitrates, intravenous heparin and beta-blockers should also be started immediately.5
After the diagnosis of TC has been established, antiplatelet agents and nitrates should be discontinued, to be replaced by supportive therapy only. Since this is a catecholamine-induced syndrome, beta-blockers should be maintained and angiotensin-converting enzyme (ACE) inhibitors should also be started until recovery of cardiac function, avoiding beta agonists and vasopressors whenever possible even in acute circulatory failure, in which mechanical circulatory support is preferred.3,5
Thus, even in cases of hypotension or cardiogenic shock, the use of intra-aortic balloon pump counterpulsation and mechanical ventilation is preferred to inotropic agents, which may be deleterious.28 When there is dynamic LV outflow tract obstruction, none of the above should be used, nor ACE inhibitors, angiotensin receptor blockers or diuretics, because of the risk of potentiation.3,29
Although TC can be triggered by emotional stimuli, it has received little attention in the psychiatric literature. Some studies have suggested the use of beta-blockers to prevent post-traumatic stress disorder and to reduce the impact of future emotional stress on the patient's functional status. In the event of acute stress, altering the autonomic activity of the heart using relaxation therapy, biofeedback, social support, meditation, hypnosis, slow breathing or yoga may be beneficial, but further research is necessary in this area.30
Diuretics are recommended in cases of heart failure (HF), and short-term anticoagulation is indicated in patients with atrial fibrillation or thrombi.31
With regard to recurrence, animal studies show that norepinephrine-induced cardiac injury is more effectively reduced by pretreatment with alpha-blockers than with beta-blockers, which is in agreement with studies reporting that prior chronic beta-blocker therapy does not appear to prevent the development of TC.32
It is also important to treat secondary causes of TC such as hyperthyroidism, although it is not clear if thyroid hormones exacerbate a patient's response to catecholamines or if it is the autoimmune nature of hyperthyroidism that precipitates the cardiomyopathy. It is also not known whether failure to correct hyperthyroidism affects the prognosis of TC, which is known to be self-limited.11
PrognosisMost patients with TC have an excellent prognosis, with an apparently benign natural history and full and relatively speedy recovery of ventricular function.32,33 Symptoms generally resolve and ECG alterations, elevation of cardiac biomarkers and wall motion abnormalities disappear in six to eight weeks, although the ECG can take years to normalize.7,34 In-hospital mortality is low (less than 2%).7,35
However, some recent studies have shown that long-term outcomes are worse than previously reported, with overall survival of 93% in follow-up. The acute systolic dysfunction found in TC, with LVEF of less than 40%, carries a high risk of acute complications such as heart failure, pulmonary edema, cardiogenic shock, atrial or ventricular arrhythmias, ventricular septal defect, free wall rupture, apical thrombus and recurrent hospitalizations.32,36
Systolic heart failure is the most common complication of TC (around 45% of cases), and requires prompt detection and treatment. Mayo Clinic researchers have developed a risk score for the development of HF, with one point for each factor at admission (age >70 years, presence of physical stressor, and LVEF <40%). The higher the score, the greater the risk of developing acute HF: the presence of one, two, and three points was associated with a 28%, 58%, and 85% risk, respectively.28 It has also been shown that patients with higher C-reactive protein and lower LVEF have a greater risk of cardiogenic shock and death.18
Another complication that is increasingly reported is cardiac rupture, which is associated with rapid clinical decline and is fatal if not surgically repaired. Risk factors for this complication include female gender, older age, persistent ST elevation and higher blood pressure and LVEF, which suggests that higher intracardiac pressures predispose to this outcome and that beta-blockers may provide protection against cardiac rupture.37
Long-term survival in patients who recover completely is similar to those in the general population of the same age and gender; recurrence is less than 10%.7,31,38 Nevertheless, long-term follow-up is recommended.8,39
Recently, some authors have reported activation of cell survival pathways in TC and have speculated that these may trigger unregulated cell growth, suggesting that TC could be a starting point for tumorigenesis.27 On the other hand, others argue that cancer can trigger TC as a result of paraneoplastic phenomena; a higher incidence of colorectal and breast cancer and melanoma has been reported in retrospective studies of TC patients.31,40 It is possible that during the process of inducing self-proliferation, cancer cells cause adrenoreceptor dysregulation affecting cardiac cells, distorting their sensitivity and resulting in TC, which would thus have a considerably less benign prognosis than hitherto believed.31 Another possibility is that a diagnosis of cancer may alter the psychic threshold for stress stimuli, so that lower stimuli may generate inappropriate increases in cardiac sympathetic nervous activity.40 However, this hypothesis does not explain why TC does not occur in all people with adenocarcinoma, nor does it take into account the real prevalence of such cancers in the age-group mainly affected by TC.31
ConclusionAlthough takotsubo cardiomyopathy is the subject of a growing body of research, the number of cases described is relatively small and much is still unknown about its pathophysiology, treatment and prognosis.8 National data have been published for some countries,13 but a large-scale registry of TC would help promote a wide-ranging study of its pathology, including molecular analysis and experimental studies, with the aim of determining its primary and secondary causes.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Nóbrega, S; Miocardiopatia Takotsubo: estado da arte. Rev Port Cardiol. 2012. http://dx.doi.org/10.1016/j.repc.2012.02.014.
