





Sarcomeric hypertrophic cardiomyopathy (HCM) is the most common genetic cause of unexplained left ventricular hypertrophy and has no specific treatment. Anderson‐Fabry disease (AFD) is rare and usually multisystemic, but occasionally expresses clinically as a predominantly cardiac phenotype mimicking HCM. We describe an illustrative case of a patient followed regularly for 25 years with a diagnosis of familial HCM and no identified sarcomeric mutations. Next‐generation sequencing analysis identified a novel pathogenic mutation in the GLA gene, leading to a diagnosis of previously unknown multisystemic AFD, with consequent implications for the patient's treatment and prognosis and familial screening.
A miocardiopatia hipertrófica sarcomérica é a causa genética mais comum da hipertrofia ventricular esquerda inexplicada e não tem tratamento específico. A doença de Anderson‐Fabry é rara, geralmente multissistémica mas, ocasionalmente, pode expressar‐se clinicamente com um fenótipo predominantemente cardíaco, imitando miocardiopatia hipertrófica. Os autores descrevem o caso ilustrativo de uma doente seguida regularmente durante 25 anos com o diagnóstico de miocardiopatia hipertrófica familiar, sem mutação sarcomérica identificada. A utilização da análise de sequenciação de nova geração identificou uma mutação patogénica nova no gene GLA, aclarando o diagnóstico oculto de doença de Anderson‐Fabry multissistémica, com as consequentes implicações terapêuticas, prognósticas e na investigação familiar.
Hypertrophic cardiomyopathy (HCM) is a common autosomal dominant disease1 limited to the heart and associated with mutations in sarcomere‐related genes. It is characterized by unexplained left ventricular hypertrophy (LVH). Its penetrance is variable and age‐related; some mutations determine the phenotype well after middle age.2,3 In 30%–40% of patients no mutations are found.4
Anderson‐Fabry disease (AFD) is a rare X‐linked lysosomal storage disease, caused by a deficiency in the enzyme alpha‐galactosidase A (alpha‐Gal A) due to mutations in the GLA gene. It leads to accumulation of globotriaosylceramide (Gb3) within lysosomes, resulting in multiorgan cell dysfunction. In its classical form it is a multisystemic disease, most frequently involving the kidneys, heart, nervous system and skin.5 Males (hemizygous) are more severely affected and usually manifest earlier than females (heterozygous).6,7 The predominant cardiac phenotype usually reflects late‐onset AFD and may be associated with GLA mutations that maintain residual enzymatic activity. Thus the cardiac variant presents unexplained LVH diagnosed in middle‐aged patients.8 Differentiating AFD from sarcomeric HCM on a clinical basis can be very difficult, but the distinction is important since specific treatment is now available for AFD (enzyme replacement therapy). Alpha‐Gal A activity in plasma or peripheral leukocytes is low in most affected men and confirms the diagnosis.9 However, in women differential diagnosis between the two conditions usually requires genetic testing.
We report the identification of a novel mutation in the GLA gene by next‐generation sequencing (NGS) analysis in a female patient followed for 25 years with a diagnosis of familial HCM.
Case report – part IIn 1987, a 34‐year‐old woman sought cardiological advice because her father (who died at 61) had been diagnosed several years before as having HCM and heart failure (HF). The woman was apparently healthy and had normal physical examination. The ECG showed sinus rhythm (SR) with a heart rate of 50 bpm and short PR interval (0.11 s) but was otherwise normal. The echocardiogram (echo) was also normal.
After ten years of annual follow‐up, at 44 years of age a diagnosis of HCM was made on the basis of an LVH pattern on ECG with significant anterolateral ST‐T strain changes (Figure 1A). One year later, an echo showed mild septal (13 mm) and lateral‐distal (16 mm) hypertrophy (Figure 2, top).

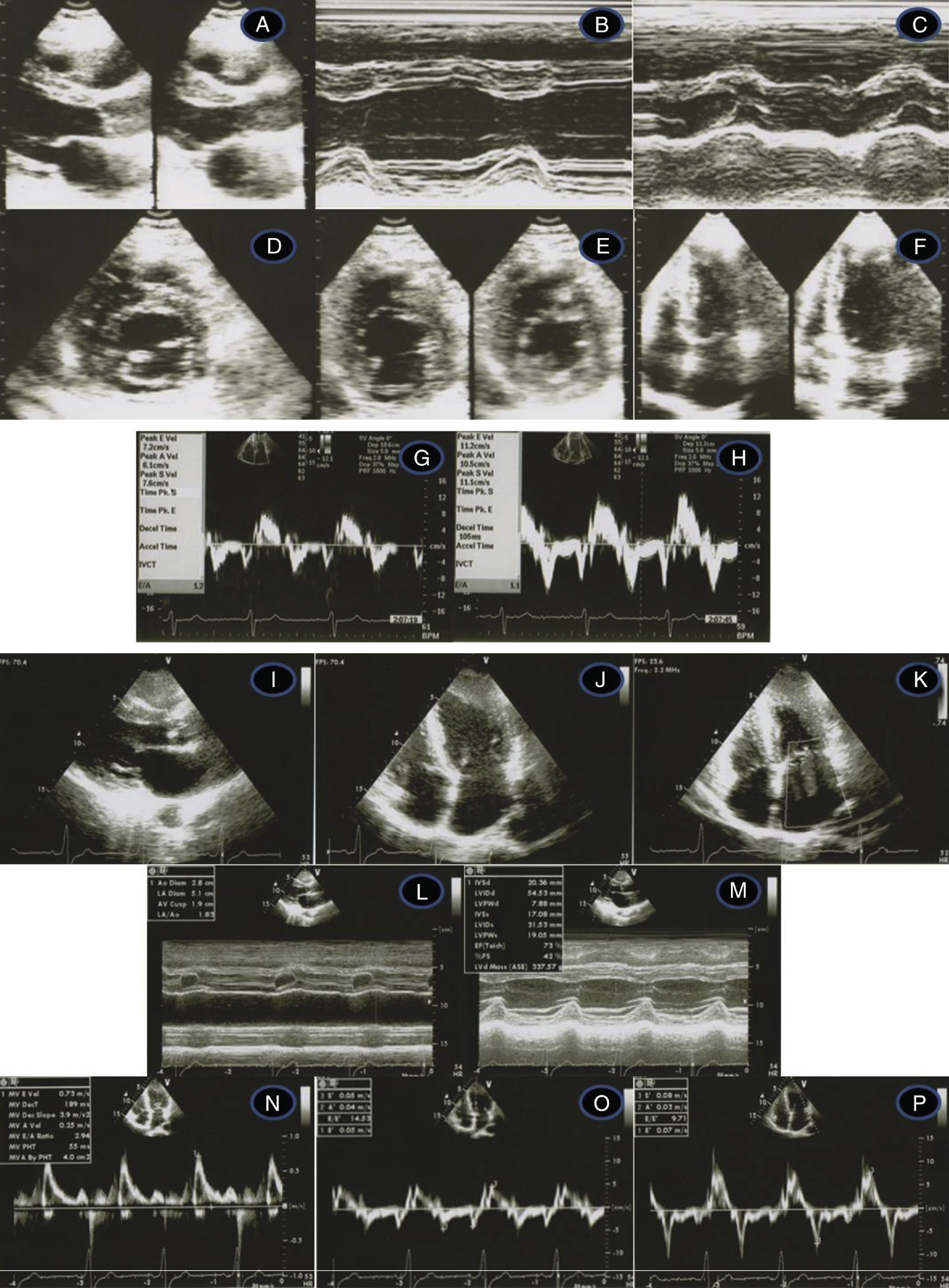
Transthoracic echocardiogram/Doppler imaging. Top (1998, age 45 years): long‐axis view showing mild septal thickening, non‐dilated and normally contracting left ventricle, and normal left atrium (A, B, C); short‐axis view at the level of the mitral leaflet tips (D) and mid‐cavity (E); apical 4‐chamber view showing slight thickening of the lateral‐apical wall (F); middle (2007, age 54 years) – normal tissue Doppler imaging at the septal (G) and lateral (H) corners of the mitral annulus; bottom (2012, age 59‐years) – thickened septum (I, J, M – 20 mm); thickened lateral‐apical wall (J); non‐dilated and normally contracting left ventricle (M); dilated left atrium, 51 mm (L); mild mitral regurgitation (K); diastolic dysfunction and affected longitudinal systolic LV function (N, O, P – see text for details).
Over the following eight years, septal thickness progressed to 17 mm, the left atrium (LA) enlarged to 44 mm and prolonged mitral inflow deceleration time (DT) of >300 ms was observed on Doppler echo, suggesting LV diastolic dysfunction. She complained of palpitations (short runs of supraventricular tachycardia were detected on Holter monitoring) but became asymptomatic after effective beta‐blocking treatment. Periodic exercise tests were normal (including exercise tolerance and blood pressure response). She had no clinical evidence of other organ involvement.
Genetic testing became possible when she was 50 years old and the six most common HCM‐associated sarcomeric genes (MYBPC3, MYH7, TNNT2, TNNI3, MYL2, and MYL3) were screened for mutations by PCR and direct sequencing of all coding regions. The results were negative.
At the age of 54 years the LVH pattern on the ECG was striking (Figure 1B), and while septal hypertrophy remained stable, apical LVH on echo was more pronounced. There was mild mitral regurgitation (MR) but mitral inflow parameters were similar. On tissue Doppler imaging (TDI), peak diastolic (E′) septal and lateral mitral annulus velocities were respectively 7.2 cm/s and 11.2 cm/s. The E/E′ ratio was <8 and systolic velocities were also normal (Figure 2, middle).
Three years later she had an episode of HF. Atrial fibrillation (AF) was diagnosed (Figure 1C) and successfully converted to SR by electrical cardioversion. Plasma NT‐proBNP was 5816 pg/ml. Echocardiographic examination showed a dilated LA and mild MR persisted. Warfarin was added to the therapy. The plasma NT‐proBNP value at discharge was 3555 pg/ml.
During the following two years (2011–2012) she remained stable although with moderate intolerance to daily exertion. Several recurrences of AF worsened her fatigue severely but were always converted to SR. Thyroid function tests were normal.
Recent echocardiographic examination (Figure 2, bottom) showed septal thickness of 20 mm, marked apical LVH, significant diastolic dysfunction (much lower E′ septal and lateral velocities, respectively 5.0 cm/s and 7.0 cm/s), higher LV filling pressures (septal E/E′ 14.5) and compromised longitudinal systolic function (septal and lateral systolic velocities of 5.0 cm/s and 8.0 cm/s) (Figure 2, bottom). The mitral inflow Doppler pattern changed to almost restrictive filling (E/A 2.94; DT 189 ms). NT‐proBNP levels remained high (1278 pg/ml), as frequently observed in HCM,10 related to the presence of LVH and to ventricular dysfunction.
A second genetic test was planned, covering a wider spectrum of genes associated with the HCM phenotype, given the need to clarify the genetic diagnosis.
Since the diagnosis of familial HCM, her first‐degree relatives have also been regularly followed. The daughter was apparently normal. The patient's only brother and her mother both had mild septal hypertrophy and a history of hypertension. Her nephew was normal.
Mutation screening and resultsIn addition to the six genes screened previously, further 13 genes associated with HCM phenotypes – ACTC1, ACTN2, CSRP3, GLA, LAMP2, MYL3, MYOZ2, NEXN, PLN, PRKAG2, TNNC1, TPM1 and TTR – were analyzed in genomic DNA by oligonucleotide‐based target capture (SureSelect, Agilent) followed by next‐generation sequencing (Illumina HiSeq2000). Sanger sequencing was used to provide data for bases with insufficient coverage, and to confirm all novel variants.
NGS detected a novel hemizygous single nucleotide exchange in the GLA gene (GenBank accession number NM_000169.2): c.187T>A (p.Cys63Ser), affecting an amino acid that is highly conserved in various species (Figure 3B). A pathogenic mutation in the same codon but with a different amino acid change (p.Cys63Tyr) has been reported by Schäfer et al.11 The change of the amino acid cysteine to tyrosine or to serine disrupts a disulfide bond of the protein that is essential for maintaining the correct structure of the protein's active site.
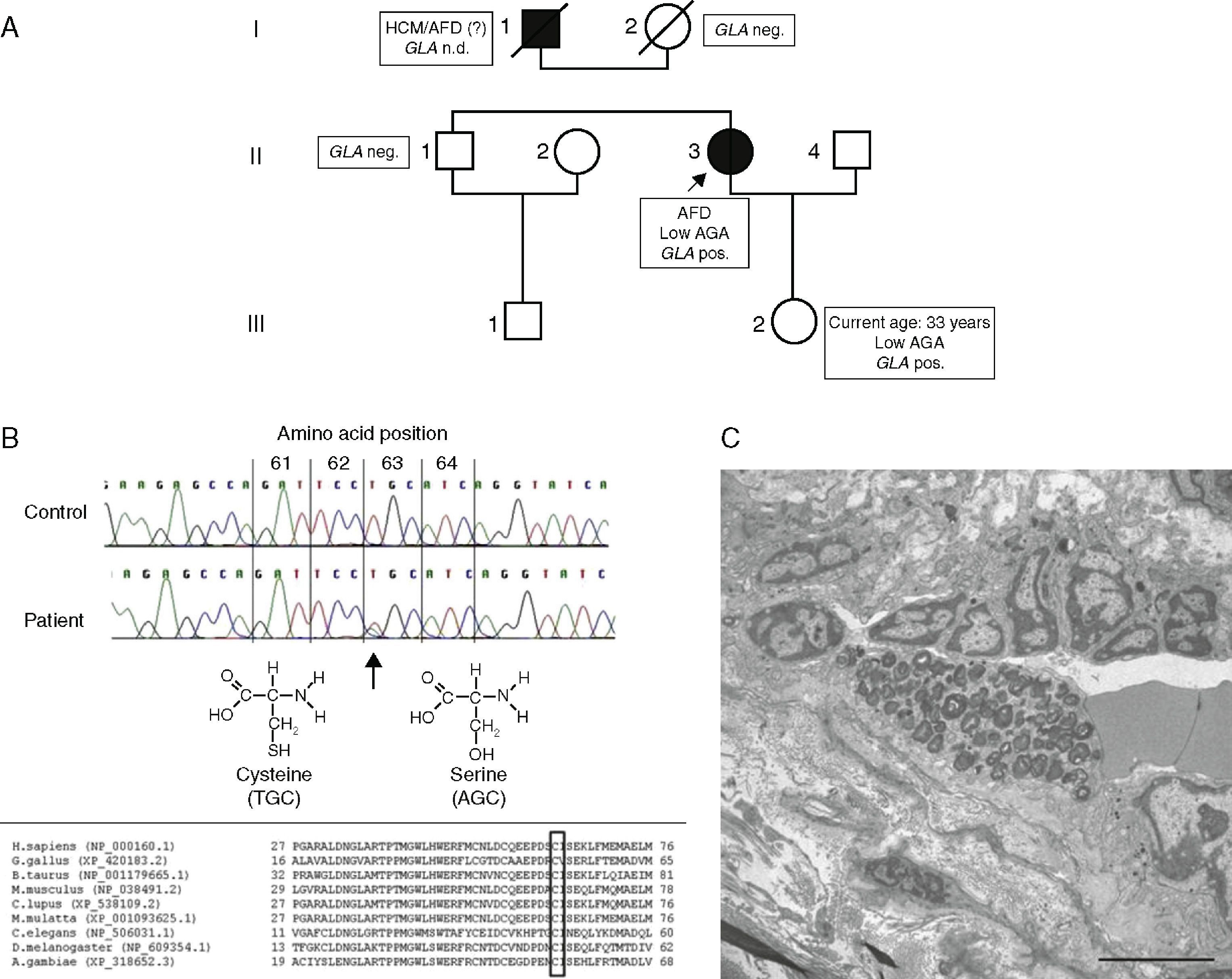
(A) Pedigree and main clinical, enzymatic and genetic findings of the family. Circles indicate females; squares, males; filled symbols, affected individuals; open symbols, unaffected individuals; slash, deceased; AFD: Anderson‐Fabry disease; AGA: alpha‐Gal A; GLA pos, carrier of GLA mutation; HCM: hypertrophic cardiomyopathy; nd: no data. The index patient is marked with an arrow; (B, top) part of nucleotide sequence of exon 1 of the GLA gene, harboring the affected amino acid. Upper row: control individual, lower row: index patient. The mutation is marked with an arrow; (B, bottom) homology analysis of amino acid sequence of alpha‐Gal in various species (http://www.ncbi.nlm.nih.gov/homologene). Affected amino acid is marked with a rectangle; (C) electron micrograph of an intradermal vessel. Note the marked abnormal storage of an endothelial cell, with multilamellar and zebra bodies, characteristic of AFD. Scale bar is 5 μm.
To assess the pathogenic impact of the novel identified variant, in silico analyses was performed using the evolutionary model program SNPs&GO (http://snps‐and‐go.biocomp.unibo.it)12 and the pathogenicity prediction program PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph/).13 Both attributed a high probability of pathogenicity to this novel mutation.
Case report – part IIAfter the genetic diagnosis of AFD, the index patient was re‐evaluated and a full organ assessment was carried out. The concentration of alpha‐Gal A in dried blood spots on filter‐paper was low (0.28 nmol/h/spot: mean reference in 120 controls: 0.64) and urine analysis showed a significant increase in Gb3 (Gb3/sphingosine 1.97).
The patient reported no neurological, cutaneous, gastrointestinal or pulmonary involvement, but the ophthalmologic examination showed cornea verticillata typical of the disease. Although the patient denied hearing loss, audiometric studies revealed cochlear involvement with sensory deficits on the right for all tested frequencies (125–8000 Hz).
She had proteinuria (317.5 mg/24 hours) and although serum creatinine was within the normal range (0.7 mg/dl), she had a mildly impaired glomerular filtration rate of 52 ml/min/m2 by the Cockcroft‐Gault equation.
A biopsy of normal skin observed by electron microscopy revealed lysosomic inclusions (“zebra bodies”) typical of AFD (Figure 3C). Brain magnetic resonance imaging (MRI) (PD, T2 and FLAIR) revealed multiple white matter lesions in the frontal and parietal regions of both hemispheres compatible with microvasculopathy. Cardiac MRI showed no areas of late gadolinium enhancement, making cardiac fibrosis unlikely. Therapy with recombinant alpha‐Gal A every two weeks was initiated.
The patient's daughter, brother and mother were screened for the p.Cys63Ser mutation, which was found in the daughter and enzyme analysis revealed reduced alpha‐Gal A activity (0.41 nmol/h/spot). The mutation was excluded in the patient's brother and mother (Figure 3A).
Considering the possible diagnosis of AFD in the father, some retrospective investigation was carried out. The patient reported that her father had been a “very sick man since his youth”. He frequently had “pain in his hands and feet” that was never investigated. However, it was not possible to rely on these data to make any diagnosis. His main problem was “heart disease” with a diagnosis of HCM since his 40s, and had been regularly followed thereafter. Available exams from a hospital discharge (admitted for “HCM and HF” at age 61) revealed AF and LVH on the ECG, “severe and diffuse LVH, dilated LA and excellent systolic function” (echo) and “severe mitral regurgitation, preserved LV systolic function and normal coronary arteries” (hemodynamic study). He was referred for mitral prosthesis implantation but committed suicide while awaiting surgery. It is worth noting that according to the hospital discharge report all laboratory parameters were in the normal range.
DiscussionAFD is rare, has a heterogeneous clinical course, especially in women,14,15 and is often not considered as a first diagnosis in the presence of cardiac isolated hypertrophy. In fact the diagnosis of AFD is particularly challenging in female patients when non‐cardiac traits are absent. In these cases the presence of LVH is usually attributed to sarcomeric HCM.
In our patient, low alpha‐Gal A activity, high urinary levels of the biomarker Gb3 and the results of the skin biopsy filled the gap between the identified mutation and the cardiac phenotypic presentation and proved that the novel mutation was pathogenic.
In retrospect, the first clue of cardiac AFD in our patient was at 34 years of age, when an ECG showed a short PR interval with no delta wave, a finding observed in up to 40% of adults with AFD.16 However, normalization of the PR interval can be expected as the patient gets older, as in our case. Only 10 years after the first observation, an LVH pattern appeared on the ECG – which is present in 18% of women carriers17 – followed by mild LVH on echocardiographic examination. In women with AFD, the progression to LVH becomes manifest between the ages of 40 and 50,18 as in this patient. Asymmetric LVH is in fact unusual in AFD, as is involvement of the apical lateral wall, as in the case presented.
Mild to moderate diastolic dysfunction is frequently observed in AFD cardiomyopathy but a restrictive pattern is uncommon.17 In advanced disease systolic heart failure may also occur. Our patient showed progressive ventricular dysfunction with a pattern of mitral inflow and diastolic TDI almost meeting the criteria for restrictive pathophysiology. This explains her very poor tolerance to AF episodes. Longitudinal systolic function was also affected.
Several studies have reported data on the prevalence of AFD in cohorts of patients with HCM, with different results.8,19–22 A screening study among 508 HCM patients found that around 1% in fact had AFD on further evaluation.19 In the ACES study,20 which included 1386 patients with unexplained LVH, the prevalence of GLA mutations was only 0.5%. Higher prevalences were found in previous smaller studies,8,21,22 while Havndrup et al.,23 in a cohort of 59 probands with HCM and no identified sarcomere gene mutations (nine genes screened), found a mutation in the GLA gene in 5%. However, in women with HCM and with no identified sarcomere mutations, the prevalence of AFD was 10% and in those older than 45 it reached 13%.
AFD is nowadays a potentially treatable cause of LVH, in contrast to sarcomeric HCM, for which no specific treatment exists. As cardiac disease is the most frequent cause of death in AFD in both genders,24,25 and as women with AFD are more likely to develop a predominantly cardiac phenotype, the clinical diagnostic algorithm suggested by Havndrup et al.23 and recently supported by other authors26 seems an appropriate approach when facing an unexplained diagnosis of HCM: when there is no clear documented family history of HCM and the proband is negative for sarcomere mutations, AFD should always be considered. Alpha‐Gal A enzyme activity in plasma or peripheral leukocytes should be assessed in males but suspected female patients should undergo gene testing as a first‐line approach.
Our patient's final diagnosis was revealed by next‐generation sequencing technologies and included a total of 19 HCM‐associated genes. NGS methods have opened a new era in routine molecular genetic diagnosis, providing reliable (with practically 100% sensitivity), rapid and cost‐effective analysis of several genes in parallel. As our case demonstrates, the application of NGS in unclear HCM cases can provide a positive result and thus reveal the exact cause of the disease. In the near future, NGS will probably become the method of choice in routine diagnosis world‐wide. If so, correct interpretation of the various novel and sometimes multiple mutations detected in a patient will demand particularly close collaboration between physicians, geneticists and the testing laboratory.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data and that all the patients included in the study received sufficient information and gave their written informed consent to participate in the study.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
The authors thank Monteiro Grilo (MD, PhD – Ophthalmology), Jorge Campos (MD, PhD – Neuroradiology), Ana Almeida (MD, PhD – Cardiac MRI), Ana Claro (MD – ORL), Paulo Filipe (MD, PhD – Dermatology), Ana Coutinho (PhD – Genetics) and Olga Azevedo (MD) for their invaluable contributions to the study of this family. The authors thank Shire and Merck Sharp & Dohme for their support in the study of this family.
Grant from the Fondo de Investigación Sanitaria, Spanish National Institute of Health Carlos III, Ref. PI10/02628, to CN.
