

The diagnosis of Marfan syndrome (MFS) depends on a multidisciplinary clinical evaluation. Molecular study to identify mutations in the FBN1 gene can establish a definitive diagnosis even with atypical or “incomplete” phenotypes and enable earlier diagnosis in asymptomatic patients.
ObjectivesThe aim of the present work was to evaluate the frequency and type of FBN1 gene mutations in a population of Marfan syndrome patients referred to a tertiary care center with cardiothoracic surgery.
MethodsOur sample included 30 individuals with MFS (from 14 families), evaluated in cardiology, rheumatology and ophthalmology consultations. In all patients, DNA was extracted from a peripheral blood sample and mutation screening of the entire coding sequence of the FBN1 gene was then performed, using the polymerase chain reaction.
ResultsWe identified 12 different mutations in the 14 families studied. Of these, only two had been previously described in the literature, while the other 10 were found to be new mutations; 36% of patients carried a Missense mutation and 50% carried a mutation leading to a premature termination codon.
ConclusionsTo the best of our knowledge this is the first genotypic description of Portuguese patients with MFS. In this study, we highlight the need for comprehensive clinical evaluation of these patients and the value of FBN1 mutation analysis in selected cases. By describing 10 new mutations, we have also helped broaden the spectrum of known FBN1 mutations associated with MFS.
O diagnóstico da Síndrome de Marfan (SM) depende fundamentalmente de uma avaliação clínica multidisciplinar. O seu diagnóstico molecular, através da identificação de mutações no gene FBN1, pode permitir estabelecer um diagnóstico definitivo mesmo perante fenótipos atípicos ou «incompletos» e reconhecer precocemente portadores assintomáticos.
ObjectivosO presente trabalho teve como objectivo principal avaliar a frequência e o tipo de mutações no gene FBN1, numa população de doentes com SM, referenciados a um centro hospitalar de cuidados terciários, com cirurgia torácica.
MétodosA nossa amostra incluiu 30 indivíduos com SM (provenientes de 14 famílias), que foram avaliados em consulta de Cardiologia, Reumatologia e oftalmologia. Em todos os casos foi efectuada a pesquisa de mutações no gene FBN1 a partir de ADN obtido de amostras de sangue periférico, utilizando a técnica de amplificação por Polymerase Chain Reaction e posterior sequenciação génica.
ResultadosIdentificámos 12 mutações distintas nas 14 famílias estudadas. Destas, apenas 2 estavam previamente descritas na literatura, sendo as restantes 10, novas mutações. Encontrámos mutações missense em 36% dos casos e mutações conduzindo à formação de codões de terminação prematura em 50% dos casos.
ConclusõesEste trabalho constituia primeira descrição de resultados de análise genotípica em doentes portugueses com SM, de que temos conhecimento. Com este trabalho, realçamos a importância de uma avaliação clínica multidisciplinar e da utilidade da pesquisa de mutações no gene FBN1 em casos seleccionados. Ao descrever 10 novas mutações, contribuímos ainda para a ampliação do espectro de variantes do gene da FBN1 associadas à SM.
The diagnosis of Marfan syndrome (MFS) depends on a multidisciplinary clinical evaluation and identification of a set of diagnostic criteria, recently revised, known as the Ghent criteria 1. The spectrum of clinical manifestations is very wide, but typically the cardiovascular, musculoskeletal, and ocular systems are involved.
Molecular study to identify mutations in the FBN1 gene can establish a definitive diagnosis even with atypical or “incomplete” phenotypes. This gene, located on chromosome 15q21.1, codes for a glycoprotein, profibrillin-1, which is then converted to fibrillin-1, a major component of extracellular microfibrils. Over 600 different mutations have been described 2, almost all unique to an affected individual or family 3.
Mutations may be found anywhere on the FBN1 gene, and may be missense or lead to a premature termination codon (PTC) 3.
Of the few genotype-phenotype correlations that have been established, the most notable is the association of neonatal MFS, the most severe form of the syndrome, with mutations in exons 24-32.
Early identification of asymptomatic carriers is desirable given the high penetrance of mutations and the morbidity and mortality associated with MFS, mainly related to the risk of aneurysm rupture and dissection of the ascending aorta 4. There are treatments that can improve the prognosis of these patients if applied in time.
There are few published data concerning the molecular diagnosis of patients with MFS in the Portuguese population.
ObjectivesThe aim of the present work was to evaluate the frequency and type of FBN1 gene mutations in a population of Marfan syndrome patients referred to a tertiary care center with cardiothoracic surgery.
MethodsIdentification of index casesPatients were identified from the hospital's database, covering a period of 20 years, between January 1989 and December 2008. This database includes all patients attending consultations and/or hospitalized in our institution.
The selection criteria were current age of 18 years or over and a diagnosis of “Marfan syndrome”.
The adult patients identified were contacted by mail or telephone and their physicians were informed that they patients were to be included in the research project.
All first-degree relatives of the index cases were invited to participate in the study.
Clinical assessmentAll participants were evaluated or re-evaluated in cardiology, rheumatology and ophthalmology consultations between October 2008 and May 2009.
Echocardiographic exams were performed by two experienced operators, using Vivid 3 (GE Healthcare) or iE33 (Philips) scanners, and measurements were made in accordance with the guidelines of the American Society of Echocardiography and the European Association of Echocardiography 5.
Images were acquired in left parasternal, apical, suprasternal and subcostal planes. Aortic root diameter was measured in parasternal long-axis view at the level of the sinuses of Valsalva and the sinotubular junction of the ascending aorta, and the diameters of the aortic arch and the descending thoracic aorta were measured in suprasternal view.
Aortic root dilatation was considered to be present when the diameter measured at the sinuses of Valsalva exceeded the upper normal limit adjusted for the patient's age and body surface area 6.
The presence of scoliosis and protrusio acetabuli was determined from X-rays of the spinal column and hip, respectively, in rheumatology consultations. The cutaneous criteria for MFS were assessed in dermatology consultations.
Respiratory system involvement was assessed through the clinical history, patients being asked about previous pneumothorax.
The presence of ectopia lentis was investigated in all patients in ophthalmology consultations.
Molecular studyDNA was extracted from a peripheral blood sample and mutation screening of the entire coding sequence of the FBN1 gene (65 exons) was then performed using the polymerase chain reaction (PCR).
If no mutations were identified by this technique, multiplex ligation-dependent probe amplification (MLPA) was used to detect gene rearrangements. All positive results were confirmed by repeating the entire analysis.
In cases of mutations not previously described in the literature (the UMD database) 2, pathogenicity was investigated by comparison with the Ensembl nucleotide polymorphism database 7 and using the PolyPhen algorithm (PolyPhen database) 8.
Statistical analysisThe statistical analysis was performed using SPSS version 16.0 (SPSS Inc., Chicago, IL, USA). Continuous variables are presented as means ± standard deviation, or as medians (interquartile range) in the case of non-normal distribution. Categorical variables are presented as frequency counts and percentages. Differences between means were assessed by the Student's t test for independent samples, except for non-normal distributions, for which the Mann-Whitney U test was used. A value of p <0.05 was used to reject the null hypothesis.
ResultsThe study population included 30 individuals with MFS from 14 unrelated families (14 index cases and 16 relatives), 57 % male, mean age 37±12 years.
Of these, 27 (90 %) presented major cardiovascular criteria; 13 (43 %) had undergone surgical aortic root replacement, 43 % had mitral valve prolapse, 23 % evidence of aortic dissection and 23 % dilatation of the descending aorta. Regarding medication, 19 patients (63 %) were taking beta-blockers, six (20 %) angiotensin-converting enzyme inhibitors, and seven (23 %) angiotensin II receptor blockers.
The prevalences of the Ghent criteria are shown in Table 1.
Prevalences of the Ghent criteria in the study population
| Ghent criteria | No. of patients/Total study population (% valid) |
| Arm span to height ratio >1.05 | 15/30 (50.0) |
| Wrist and thumb signs | 12/30 (40.0) |
| Pectus carinatum | 8/30 (26.7) |
| Pectus excavatum requiring surgery - not requiring surgery | 1/30 (3.3)-6/30 (20.0) |
| Pes planus | 17/28 (60.7) |
| Protrusio acetabuli | 17/19 (89.5) |
| Scoliosis of >20° | 13/29 (44.8) |
| Joint hypermobility | 21/30 (70.0) |
| Facial abnormalities (micrognathia, malar hypoplasia, arched palate, crowding of teeth) | 28/30 (93.3) |
| Major musculoskeletal criteria | 16/25 (64.0) |
| Musculoskeletal involvement (without major criteria) | 14/30 (46.7) |
| Dilatation of the ascending aorta (not requiring surgery) - previous ascending aorta surgery | 12/30 (40.0)-12/30 (40.0) |
| Dissection of the ascending aorta | 7/30 (23.3) |
| Dilatation of the descending aorta | 7/30 (23.3) |
| Mitral valve prolapse (with or without surgery) | 13/30 (43.3) |
| Dilatation of the main pulmonary artery | 2/30 (6.7) |
| Major cardiovascular criteria | 27/30 (90.0) |
| Cardiovascular involvement (without major criteria) | 2/30 (6.7) |
| Striae | 17/30 (43.3) |
| Hernia | 4/30 (13.3) |
| Cutaneous involvement | 19/30 (63.3) |
| Spontaneous pneumothorax | 4/30 (13.3) |
| Apical blebs | 3/30 (10.0) |
| Pulmonary involvement | 4/30 (13.3) |
| Major ocular criteria (ectopia lentis with or without surgery) | 5/19 (26.3) |
| Family history or FBN1 mutation | 28/30 (93.3) |
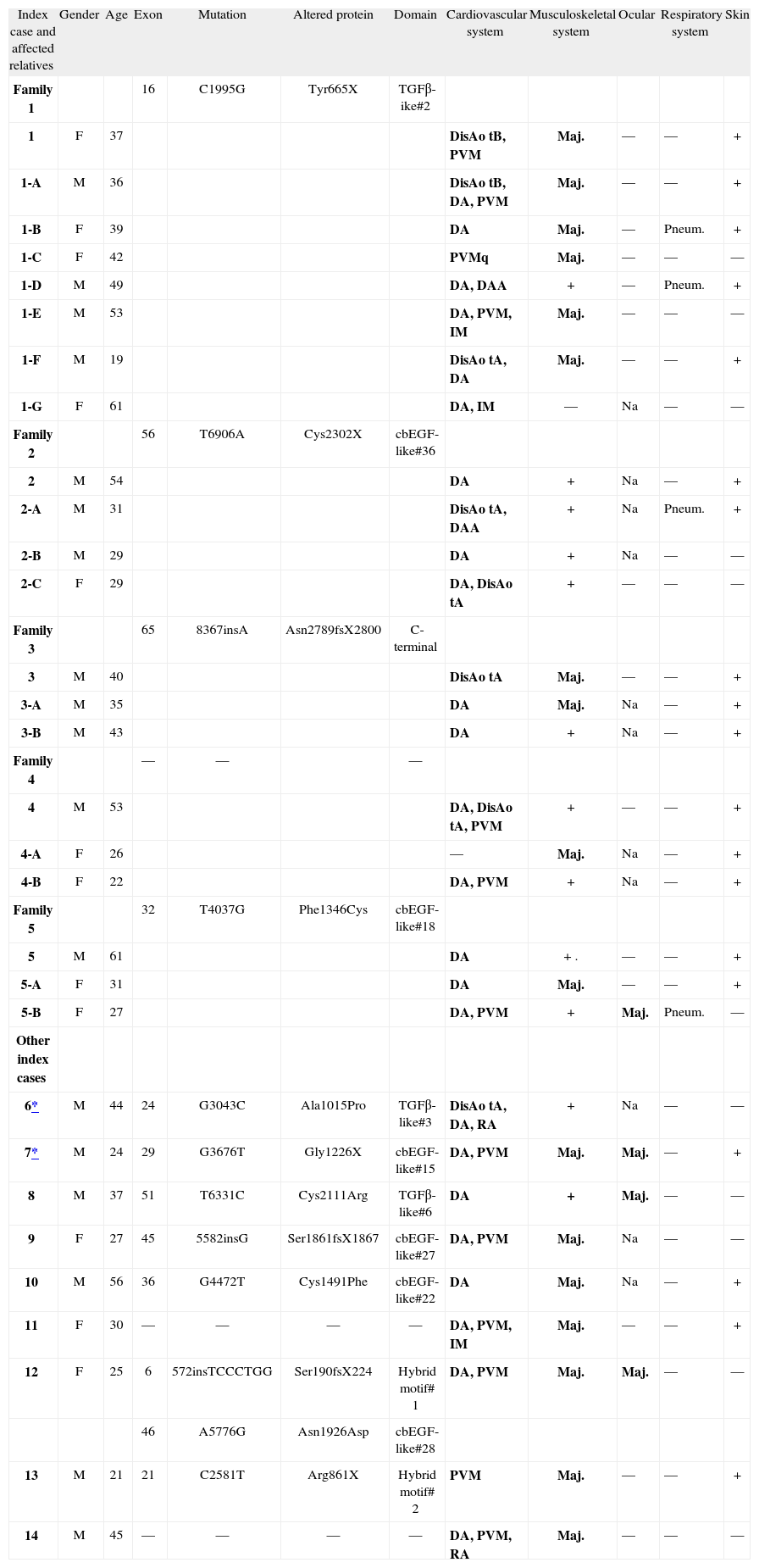
Genetic study was performed in the 14 index cases and in 22 relatives (16 with positive genetic test and 8 with no FBN1 mutation identified). The results of molecular study and their correlation with the presence of MFS criteria are shown in Tabl 2.
Molecular and clinical characteristics of 14 index cases and 16 relatives who fulfilled the Ghent criteria for Marfan syndrome
| Index case and affected relatives | Gender | Age | Exon | Mutation | Altered protein | Domain | Cardiovascular system | Musculoskeletal system | Ocular | Respiratory system | Skin |
| Family 1 | 16 | C1995G | Tyr665X | TGFβ-ike#2 | |||||||
| 1 | F | 37 | DisAo tB, PVM | Maj. | — | — | + | ||||
| 1-A | M | 36 | DisAo tB, DA, PVM | Maj. | — | — | + | ||||
| 1-B | F | 39 | DA | Maj. | — | Pneum. | + | ||||
| 1-C | F | 42 | PVMq | Maj. | — | — | — | ||||
| 1-D | M | 49 | DA, DAA | + | — | Pneum. | + | ||||
| 1-E | M | 53 | DA, PVM, IM | Maj. | — | — | — | ||||
| 1-F | M | 19 | DisAo tA, DA | Maj. | — | — | + | ||||
| 1-G | F | 61 | DA, IM | — | Na | — | — | ||||
| Family 2 | 56 | T6906A | Cys2302X | cbEGF-like#36 | |||||||
| 2 | M | 54 | DA | + | Na | — | + | ||||
| 2-A | M | 31 | DisAo tA, DAA | + | Na | Pneum. | + | ||||
| 2-B | M | 29 | DA | + | Na | — | — | ||||
| 2-C | F | 29 | DA, DisAo tA | + | — | — | — | ||||
| Family 3 | 65 | 8367insA | Asn2789fsX2800 | C-terminal | |||||||
| 3 | M | 40 | DisAo tA | Maj. | — | — | + | ||||
| 3-A | M | 35 | DA | Maj. | Na | — | + | ||||
| 3-B | M | 43 | DA | + | Na | — | + | ||||
| Family 4 | — | — | — | ||||||||
| 4 | M | 53 | DA, DisAo tA, PVM | + | — | — | + | ||||
| 4-A | F | 26 | — | Maj. | Na | — | + | ||||
| 4-B | F | 22 | DA, PVM | + | Na | — | + | ||||
| Family 5 | 32 | T4037G | Phe1346Cys | cbEGF-like#18 | |||||||
| 5 | M | 61 | DA | + . | — | — | + | ||||
| 5-A | F | 31 | DA | Maj. | — | — | + | ||||
| 5-B | F | 27 | DA, PVM | + | Maj. | Pneum. | — | ||||
| Other index cases | |||||||||||
| 6* | M | 44 | 24 | G3043C | Ala1015Pro | TGFβ-like#3 | DisAo tA, DA, RA | + | Na | — | — |
| 7* | M | 24 | 29 | G3676T | Gly1226X | cbEGF-like#15 | DA, PVM | Maj. | Maj. | — | + |
| 8 | M | 37 | 51 | T6331C | Cys2111Arg | TGFβ-like#6 | DA | + | Maj. | — | — |
| 9 | F | 27 | 45 | 5582insG | Ser1861fsX1867 | cbEGF-like#27 | DA, PVM | Maj. | Na | — | — |
| 10 | M | 56 | 36 | G4472T | Cys1491Phe | cbEGF-like#22 | DA | Maj. | Na | — | + |
| 11 | F | 30 | — | — | — | — | DA, PVM, IM | Maj. | — | — | + |
| 12 | F | 25 | 6 | 572insTCCCTGG | Ser190fsX224 | Hybrid motif# 1 | DA, PVM | Maj. | Maj. | — | — |
| 46 | A5776G | Asn1926Asp | cbEGF-like#28 | ||||||||
| 13 | M | 21 | 21 | C2581T | Arg861X | Hybrid motif# 2 | PVM | Maj. | — | — | + |
| 14 | M | 45 | — | — | — | — | DA, PVM, RA | Maj. | — | — | — |
Major criteria appear in bold type.
: patients with family history of MFS; —: system not involved; +: system involved; AAD: abdominal aortic dilatation; AD: aortic dilatation; AoDis: aortic dissection type A; AoDis B: aortic dissection type B; AR: aortic regurgitation; F: female; M: male; Maj: major criteria; MVP: mitral valve prolapse; MR: mitral regurgitation; NA: not applicable; Pneum.: pneumothorax.
Twelve different mutations were identified in the 14 families: ten new and two known (cases 8 and 13, mutations Cys2111Arg 2 and Arg861X 9). One patient had two different mutations. The mutations were identified in functionally distinct domains: three in TGF-β-like domains, six in cbEGF-like domains, two in hybrid domains, and one in the terminal COOH region.
In three index cases that fulfilled the Ghent criteria, no FBN1 mutation was found.
DiscussionTo the best of our knowledge this is the first genotypic description of Portuguese patients with MFS.
The percentage of patients fulfilling the Ghent criteria in whom mutations were detected was slightly lower than in the literature 10,11 (79 % vs. 90 %, respectively), but this may be due to the wide confidence interval (64.4 %-93.6 %) resulting from the small sample size.
The high frequency of new mutations (10 out of 12) supports current thinking that most MFS patients are carriers of a genetic variant unique to their family.
Interestingly, the type and distribution of mutations in our patients were significantly different from those in published data 9,12. We found 36 % missense mutations and 50 % leading to a premature termination codon. As expected, most carriers of a PTC mutation did not present ectopia lentis 9. In the literature, missense mutations account for two-thirds of FBN1 mutations, most of which affect cysteine residues associated with cbEGF units and thus interfere with disulfide bonds and protein stability. Individuals with such mutations are more likely to have ectopia lentis 12. The rest usually have PTC mutations, leading to the formation of incomplete proteins that interfere with normal protein polymerization (dominant negative effect) 13 or result in functionally inactive proteins through RNA degradation, reducing the total quantity of fibrillin-1 (haploinsufficiency) 10,14. Individuals with these mutations tend to present more severe musculoskeletal alterations, particularly joint hypermobility, greater propensity for aortic dissection 9,15, and a significantly lower risk for ocular involvement 9, although there is no direct relationship between phenotype severity and the quantity of mutant transcript.
Our results may have been influenced by selection bias. As the series is from a tertiary care center, a large proportion of the patients fulfilled the Ghent criteria (both cardiovascular and musculoskeletal) and therefore nonsense mutations would be likely to be more prevalent 9,12.
The fact that we discovered two different mutations in one patient highlights the value of complete sequencing of the FBN1 gene, particularly in certain patients. The few cases in the literature of compound heterozygosity in MFS 16,17 are characterized by a more severe phenotype. Multiple mutations may also contribute to phenotypic variability within families.
We did not identify any asymptomatic carriers (those with FBN1 gene mutations but no major clinical manifestations). However, screening of relatives revealed individuals with mild morphological abnormalities that were insufficient for a diagnosis of MFS but for whom we were then able to provide appropriate clinical monitoring and genetic counseling. Excluding MFS in other relatives, meanwhile, meant that they did not require periodic clinical assessments or imaging studies.
In the three cases in which no mutation was found, it is possible that a mutation existed in the promoter or non-coding intronic regions of FBN1, or in large gene rearrangements undetectable by PCR; there may also have been mutations in other genes, particularly those coding for TGF-β type I and II receptors (TGF-βI and TGF-βII) 18.
ConclusionsIn this study we have highlighted the need for multidisciplinary clinical evaluation of MFS patients and their relatives.
By describing 10 new mutations, we have broadened the spectrum of known FBN1 mutations associated with MFS.
A better understanding of the etiopathology of Marfan syndrome, including molecular study, may lead to the discovery of new prognostic markers and earlier identification of candidates for therapeutic interventions 9.
FundingThis work was funded by a grant from the Research Center of Hospital de S. Joao, Porto, Portugal.
Conflicts of interestThe authors have no conflicts of interest to declare.
