






Left ventricular noncompaction (LVNC) is a genetically heterogeneous cardiomyopathy, with familial and sporadic forms, but genetic testing only identifies a pathogenic mutation in a minority of cases. The main complications are heart failure, embolism and dysrhythmias. Herein we report a familial case of LVNC associated with a mutation in the MYH7 gene and review the literature regarding controversies in LVNC. A 50-year-old woman was referred to the cardiology clinic for palpitations. She underwent echocardiography and cardiac magnetic resonance imaging that revealed mild left ventricular systolic dysfunction and LVNC criteria. She had several episodes of non-sustained ventricular tachycardia and received an implantable cardioverter-defibrillator (ICD). Genetic testing revealed the c.1003G>C (p.Ala335Pro) mutation in the MYH7 gene. Familial screening showed clear genotype-phenotype cosegregation, which provided strong evidence for the pathogenic role of this mutation. To the best of our knowledge, this is the first report of LVNC associated with the p.Ala335Pro mutation in the MYH7 gene. This mutation has been described in hypertrophic cardiomyopathy, suggesting that the same pathogenic sarcomere mutation may be associated with different cardiomyopathies. This case also highlights the current difficulties regarding decisions on ICD implantation for primary prevention of sudden cardiac death in LVNC.
A não compactação do ventrículo esquerdo (LVNC) é uma cardiomiopatia geneticamente heterogénea, com formas familiares e esporádicas descritas. No entanto, o estudo genético permite identificar uma mutação patogénica numa minoria de casos. Neste trabalho, reportamos um caso familiar de não compactação do ventrículo esquerdo associada a uma mutação no gene MYH7 e apresentamos uma revisão das principais controvérsias na LVNC.
Mulher de 50 anos, referenciada para a consulta de cardiologia por palpitações. O ecocardiograma transtorácico e a ressonância magnética cardíaca revelaram a presença de disfunção ventricular esquerda ligeira e de critérios de LVNC. Após documentação de episódios de taquicardia ventricular não sustentada foi implantado um cardiodesfibrilhador implantável (CDI). O teste genético revelou a presença da mutação c.1003G>C (p.Ala335Pro) no gene MYH7. O rastreio familiar mostrou uma clara segregação genótipo-fenótipo, o que suporta a patogenicidade desta mutação.
Este é o primeiro caso de LVNC em associação com a mutação p.Ala335Pro no gene MYH7. Esta mutação já se encontrava previamente descrita em casos de cardiomiopatia hipertrófica, o que suporta que a mesma mutação sarcomérica pode expressar-se com diferentes fenótipos. Este caso destaca ainda as dificuldades atuais no que concerne à estratificação do risco arrítmico nos doentes com LVNC, mais concretamente para decisão de implantação de CDI em prevenção primária.
Left ventricular noncompaction (LVNC) was first described by Feldt et al. in 1969 in an autopsy study.1 It was characterized as a cardiac abnormality with a two-layered ventricular myocardium composed of a thinner compacted epicardial layer and a thicker hypertrabeculated endocardial layer.2 Prominent left ventricular (LV) trabeculae are typically located distal to the papillary muscles and the deep intertrabecular recesses communicate with the ventricular cavity.2
The prevalence of LVNC varies considerably between studies and its true prevalence is unknown. Formerly considered a very rare disease, it is now more frequently recognized, with a reported prevalence of between 0.014 and 1.3% in echocardiography laboratory series.2
The diagnosis of LVNC is still controversial and the best diagnostic method and criteria are a matter of debate. Diagnosis at autopsy is made when three or more trabeculations are present and anatomopathological findings are still the gold standard. In clinical practice, diagnosis is usually based on cardiac imaging, most commonly transthoracic echocardiography and cardiac magnetic resonance imaging (MRI)3–7 (Table 1).
Diagnostic criteria of left ventricular noncompaction.
| Study | Diagnostic criteria | Phase of cardiac cycle | Echocardiographic window |
|---|---|---|---|
| Jenni et al.3 | Two-layered myocardium (thin compacted epicardial layer and thicker noncompacted endocardial layer)Ratio of thickness of noncompacted to compacted layers>2Absence of other congenital cardiac abnormalitiesColor Doppler evidence of deep intertrabecular recesses filled with blood from the left ventricular cavity. Predominant location of the trabeculae in mid-lateral, mid-inferior and apical left ventricular segments | End-systole | Short-axis view |
| Chin et al.4 | Prominent trabeculae and deep recessesX/Y ratio <0.5, X being the distance from recesses to epicardium and Y the distance between peak of the trabeculae and epicardium | End-diastole | Parasternal short-axis view |
| Stöllberger et al.5 | More than three trabeculations protruding from the left ventricular wall, located apically to the papillary musclesTrabeculations present the same echogenicity as the myocardium and synchronous movement with ventricular contractionsPerfusion of the intertrabecular recesses from the left ventricular cavity | End-diastole | Apical 4-chamber view |
| Petersen et al.6 | Ratio between noncompacted and compacted layers >2.3 | End-diastole | Any of the three long-axis views |
| Jacquier et al.7 | Trabeculated left ventricular mass greater than 20% of total left ventricular mass | End-diastole | N/A |
The clinical spectrum of the disease is variable, ranging from asymptomatic to full-blown disease with serious complications including the classic triad of heart failure, thromboembolism or ventricular arrhythmias.1
The prognosis of LVNC was initially described as very poor with considerable morbidity and mortality,8 but was later found to be better than previously reported.9 Improved quality of imaging techniques and increased awareness of the disease among physicians led to the recognition of milder forms of LVNC as well as earlier diagnosis and treatment and the possibility of improving its prognosis. At the same time, it also enabled the detection of normal variants of LV hypertrabeculation that, although fulfilling the current controversial diagnostic criteria of LVNC, may have no clinical significance.10
The etiology of the disease remains unclear. Congenital cases of LVNC are thought to result from an arrest in compaction of myocardial fibers during fetal development.1 However, the disease is not always present at birth; acquired forms have also been described and may become detectable as late as in adulthood, sometimes in association with other diseases, such as neuromuscular disorders.1 LVNC is a genetically heterogeneous cardiomyopathy with sporadic and familial forms but genotyping only identifies a mutation in about 20% of cases.1,2
Herein we present a familial case of LVNC associated with a missense mutation in the MYH7 gene (c.1003G>C, p.Ala335Pro) that had previously been described in hypertrophic cardiomyopathy, reinforcing the notion that the same sarcomere mutation may be associated with different cardiomyopathy phenotypes.
Case reportA 50-year-old female was referred to the cardiology clinic because of frequent spontaneous palpitations, with a duration of a few seconds and no relation with effort, and exercise dyspnea (New York Heart Association functional class II). She denied chest pain, pre-syncope or syncope. She had no history of previous disease, substance abuse or regular medication. The physical exam and the electrocardiogram were unremarkable.
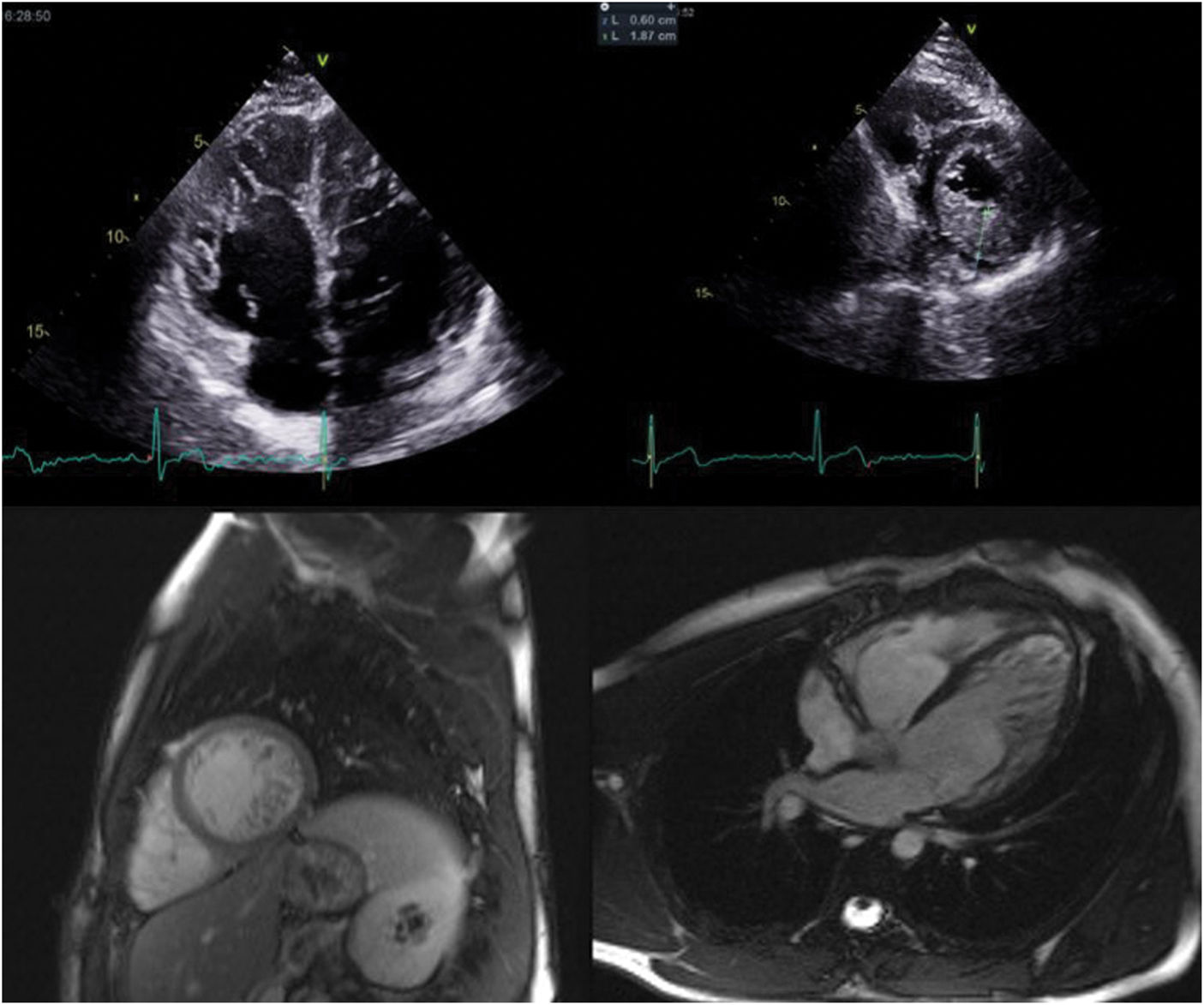
The transthoracic echocardiogram revealed LV myocardial hypertrabeculation with deep intertrabecular recesses communicating with the LV cavity as shown by color Doppler, meeting the LVNC diagnostic criteria of Jenni et al. (ratio of noncompacted to compacted layer thickness in end-systole in parasternal short-axis view: 2.5), Chin et al. (ratio of distance from the epicardium to the intertrabecular recess to the distance from the epicardium to the peak of the trabeculae in apical 4-chamber view: 0.3) and Stöllberger et al. (more than three trabeculae distal to the papillary muscles in apical 4-chamber view) (Figure 1). LV ejection fraction was 50%.

Cardiac MRI corroborated the echocardiographic findings, supporting the diagnosis of LVNC based on Petersen et al.’s criteria (ratio of noncompacted to compacted layer thickness in end-diastole: 4).
Twenty-four-hour Holter monitoring revealed sinus rhythm and 560 supraventricular and 82 premature ventricular contractions. Considering these findings and because the patient was very symptomatic and anxious, we decided to initiate medication with a beta-blocker and a benzodiazepine. Although therapeutic adhesion was confirmed, the patient did not improve and continued to report frequent palpitations, although without reporting syncope. Considering the arrhythmic risk associated with LVNC, particularly in the presence of LV systolic dysfunction, an implantable loop recorder was implanted.
Genetic testing was performed by next-generation sequencing of a panel of genes that included LDB3, TAZ, LMNA/C, MYBPC3, TNNT2, ACTC1, TPM1, CSRP3, TCAP, SGCD and PLN. We found the variant c.1003G>C (p.Ala335Pro) in exon 12 of the MYH7 gene, which was predicted as relevant by in-silico models (PolyPhen-2: probably damaging, SIFT: deleterious, Mutation Taster: damaging), but had never been described in LVNC. Interestingly, it had been previously described as a genetic variant of unknown significance in a single case of hypertrophic cardiomyopathy.11
A few weeks later, the implantable loop recorder revealed episodes of non-sustained ventricular tachycardia (VT) and the patient received an implanted cardioverter-defibrillator (ICD).
The family pedigree (Figure 2) revealed that both the patient's parents were already deceased, the father from dementia and the mother from cancer. The patient had nine siblings, including one sister who died in childhood of unknown cardiac disease and one brother who died in infancy of unknown cause. Two other brothers were deceased, one from pneumonia in infancy and one from cancer at 27 years of age. Her 49-year-old brother underwent transthoracic echocardiography, which was normal. Her 47- and 41-year-old sisters also underwent echocardiographic screening (Figure 3) and both exams resulted in a diagnosis of LVNC (Table 2). They also underwent MRI which corroborated these findings (Figure 3). The 41-year-old sister has an eight-year-old son who was also diagnosed with LVNC by echocardiographic screening. Her 28-year-old daughter has a normal echocardiogram and her 22-year-old son had pronounced LVNC with right ventricular involvement on the echocardiogram as well as on cardiac MRI (Figure 4). He also received an ICD due to non-sustained VT.
 and non-affected relatives (white symbols). The arrow indicates the proband. In this family, there is complete cosegregation between the genotype (mutation p.Ala335Pro in the MYH7 gene) and the left ventricular noncompaction phenotype. M: months; Y: years.")
Family pedigree showing affected relatives (dark symbols) and non-affected relatives (white symbols). The arrow indicates the proband. In this family, there is complete cosegregation between the genotype (mutation p.Ala335Pro in the MYH7 gene) and the left ventricular noncompaction phenotype. M: months; Y: years.
 and cardiac magnetic resonance imaging (right) from the 47-year-old sister (top) and the 41-year-old sister (bottom) of the index patient, both fulfilling the Stöllberger and Petersen diagnostic criteria of left ventricular noncompaction.")
Summary of the clinical and imaging findings of affected relatives.
| Relationship and age | Clinical findings | Echocardiogram | MRI |
|---|---|---|---|
| Sister, 47 years | Effort dyspneaPalpitations | Normal LV functionHypertrabeculated myocardium meeting the Chin, Jenni and Stöllberger criteria of LVNC | Petersen criteria of LVNC (ratio of thickness of noncompacted/compacted layers: 3.5)LVEF: 59%No late enhancement |
| Sister, 41 years | Asymptomatic | Normal LV functionHypertrabeculated myocardium meeting the Chin, Jenni and Stöllberger criteria of LVNC | Petersen criteria of LVNC (ratio of thickness of noncompacted/compacted layers: 2.8)LVEF: 64%No late enhancement |
| Son, 22 years | PalpitationsNon-sustained VT | Normal LV functionHypertrabeculated myocardium meeting the Chin, Jenni and Stöllberger criteria of LVNC | Petersen criteria of LVNC (ratio of thickness of noncompacted/compacted layers reported as >2.3)LVEF: 53%No late enhancement |
| Nephew, 8 years | Asymptomatic | Normal LV functionHypertrabeculated myocardium meeting the Chin, Jenni and Stöllberger criteria of LVNC | N/A |
LV: left ventricular; LVEF: left ventricular ejection fraction; MRI: magnetic resonance imaging.
 2, clearly fulfilling the diagnostic criteria of Jenni et al. for left ventricular noncompaction (LVNC) (top right); cardiac magnetic resonance images in short-axis view of the same patient showing hypertrabeculation (bottom left) and ratio of the thickness of noncompacted to compacted layers >2.3 in 4-chamber view, fulfilling Petersen et al.’s criteria of LVNC (bottom right).' title='Transthoracic echocardiogram in 4-chamber view of the index patient's son showing biventricular involvement (top left) and a ratio of thickness of noncompacted to compacted layers >2, clearly fulfilling the diagnostic criteria of Jenni et al. for left ventricular noncompaction (LVNC) (top right); cardiac magnetic resonance images in short-axis view of the same patient showing hypertrabeculation (bottom left) and ratio of the thickness of noncompacted to compacted layers >2.3 in 4-chamber view, fulfilling Petersen et al.’s criteria of LVNC (bottom right).'/>
2, clearly fulfilling the diagnostic criteria of Jenni et al. for left ventricular noncompaction (LVNC) (top right); cardiac magnetic resonance images in short-axis view of the same patient showing hypertrabeculation (bottom left) and ratio of the thickness of noncompacted to compacted layers >2.3 in 4-chamber view, fulfilling Petersen et al.’s criteria of LVNC (bottom right).' title='Transthoracic echocardiogram in 4-chamber view of the index patient's son showing biventricular involvement (top left) and a ratio of thickness of noncompacted to compacted layers >2, clearly fulfilling the diagnostic criteria of Jenni et al. for left ventricular noncompaction (LVNC) (top right); cardiac magnetic resonance images in short-axis view of the same patient showing hypertrabeculation (bottom left) and ratio of the thickness of noncompacted to compacted layers >2.3 in 4-chamber view, fulfilling Petersen et al.’s criteria of LVNC (bottom right).'/>Transthoracic echocardiogram in 4-chamber view of the index patient's son showing biventricular involvement (top left) and a ratio of thickness of noncompacted to compacted layers >2, clearly fulfilling the diagnostic criteria of Jenni et al. for left ventricular noncompaction (LVNC) (top right); cardiac magnetic resonance images in short-axis view of the same patient showing hypertrabeculation (bottom left) and ratio of the thickness of noncompacted to compacted layers >2.3 in 4-chamber view, fulfilling Petersen et al.’s criteria of LVNC (bottom right).
The genetic variant p.Ala335Pro was found exclusively and in all the relatives with LVNC, thus segregating with the disease. Non-affected family members tested negative for the mutation.
DiscussionThis case report describes a family with LVNC associated with the p.Ala335Pro mutation in the MYH7 gene.
In this case, the diagnosis of LVNC was irrefutable, since it was based on multimodality imaging and all patients fulfilled several diagnostic criteria on both echocardiography and cardiac MRI. However, the diagnosis of LVNC is frequently challenging, as the diagnostic criteria present several limitations. Kohli et al., in a study including 60 adult patients referred to a heart failure clinic and 60 healthy controls, reported that 47 patients and five controls (including four individuals of African origin) fulfilled one or more echocardiographic criteria for LVNC. In this study, 78.7% of the patients with heart failure fulfilled the diagnostic criteria suggested by Chin et al., 63.8% fulfilled Jenni et al.’s criteria and 53.2% fulfilled the criteria suggested by Stöllberger et al., but an overlap of all three diagnostic approaches was only found in 29.8% of the patients.12 This poor correlation between the three echocardiographic definitions is due to the heterogeneity of their criteria: images are collected in different planes and at different points of the cardiac cycle, and the morphological descriptions of the abnormal trabeculations differ.
Although the commercially available gene panel for LVNC at the time genetic testing of the proband was performed did not include genes that are currently recognized as relevant in the dilated cardiomyopathy/LVNC spectrum, such as TTN and BAG3, LVNC was found in association with a genetic variant of the MYH7 gene. MYH7 codes for the cardiac beta-myosin heavy chain. Myosin is a motor protein that, together with actin, constitutes the fundamental contractile unit of myocytes. Mutations in this gene are commonly present in hypertrophic cardiomyopathy,13 but have also been described in association with dilated cardiomyopathy14 and myopathy.15 In LVNC, at least 30 genetic variants of the MYH7 gene have been found, although their exact role remains unclear.16 The variant p.Ala335Pro had never been described in LVNC, although it has been reported in a single case of hypertrophic cardiomyopathy, with unknown significance.11 This variant affects a conserved amino acid in the protein and several in-silico models (PolyPhen-2, SIFT, and Mutation Taster) predict it as functionally relevant. Furthermore, there was perfect genotype-phenotype cosegregation, which strongly argues in favor of the pathogenic role of this MYH7 mutation in the development of LVNC. LVNC is a genetically heterogeneous disorder that has been associated with a variety of mutations in different genes encoding mitochondrial, cytoskeletal, Z-line and sarcomere proteins.10 Moreover, some mutations have been described in patients with LVNC as well as in patients with other phenotypes, such as hypertrophic or dilated cardiomyopathy.1,10 These observations suggest that a pathogenic mutation in the same gene results in different trabecular remodeling or maladaptive remodeling of the embryonic myocardium, which can cause different phenotypes and cardiomyopathies. On the other hand, LVNC may be found in association with neuromuscular diseases, genetic syndromes and congenital abnormalities, such as Ebstein anomaly, pulmonary atresia, aortic stenosis and ventricular septal defect.1 Therefore, it is controversial whether LVNC is a distinct nosological entity or rather a morphological trait of different diseases.
Familial recurrence is not well defined and varies widely between studies, ranging from 18 to 50% of cases, mainly due to the limitations of their retrospective designs.2 Autosomal dominant inheritance seems to be more common than X-linked inheritance, although autosomal recessive inheritance is also possible.2 The high rate of familial transmission means that family screening is essential. In this clinical case, LVNC was diagnosed in four family members, suggesting an autosomal dominant or X-linked trait.
The clinical spectrum of LVNC is very wide, ranging from asymptomatic individuals with incidental diagnosis to catastrophic embolic events, ventricular arrhythmias and heart failure.1 In a Swiss study by Oechslin et al.,8 35% of the patients suffered early death, 24% had thromboembolic events, 53% required hospitalization for congestive heart failure, 41% had ventricular tachyarrhythmias, 12% required ICDs, and 12% required heart transplantation over 44 months of follow-up. Studies performed more recently have reported a significantly lower rate of cardiac complications and death. In a British cohort comprising 45 patients consecutively identified at a referral center for cardiomyopathy, 30 patients (66%) had left ventricular dilatation and impaired systolic function, but only nine (20%) had non-sustained VT on 24-hour Holter monitoring, two (4%) had thromboembolic events, and the mean survival from death or heart transplantation was 97% at 46 months.9
The clinical decision on ICD implantation for the proband and her son in this case was difficult, due to the lack of evidence on which to base risk stratification of sudden death in primary prevention of patients with LVNC. In this family, the index case reported a family history of sudden death in one brother and one sister in infancy. Although the cause of death and diagnosis of LVNC cannot be established in these children, we can speculate that a major embolic event or ventricular dysrhythmia was the cause. In the index case and her son, symptomatic palpitations associated with non-sustained VT were a concern, leading to the implantation of ICDs. Current guidelines for prevention of sudden death recommend an ICD in the context of secondary prevention, based on a history of cardiac arrest in ventricular fibrillation or tachycardia or the presence of documented sustained VT, and for primary prevention, based on the existence of LV systolic dysfunction.17 Other documents on LVNC suggest ICD implantation in those presenting with syncope, or with symptomatic ventricular dysrhythmias or severely impaired LV systolic function.2 In a study of 44 patients diagnosed with LVNC, 75% of patients received an ICD for secondary or primary prevention in those with LV systolic dysfunction. During a follow-up of 26 months, appropriate shocks were recorded in 33% of the patients with an ICD for secondary prevention and in 13% of those with an ICD for primary prevention.18 This relatively high rate of shocks for sustained VT is corroborated in another study19 and confirms the high risk of sudden death faced by LVNC patients. Interestingly, in this cohort of 44 patients, LV dysfunction or dilatation were not frequent in the secondary prevention group, being present in only four out of 12 patients, which supports our concern that severity of LV dysfunction cannot be relied on for risk stratification of sudden cardiac death.
In the same study, in the group with ICDs for secondary prevention, there was a family history of sudden cardiac death in as many cases as of left ventricular dysfunction (four patients). Also, in two patients the only self-reported risk factor was syncope.18 These findings suggest there is much more to consider besides systolic dysfunction when assessing patients for ICD implantation in the primary prevention setting. Twenty-four-hour Holter showed more non-sustained VT and more premature ventricular contractions in the ICD recipients than in other patients, which reinforces its value in risk stratification.18 Bennett and Freudenberger also state that it is reasonable to take into consideration the degree of late gadolinium enhancement on MRI since this is an indication of more severe disease.20 All these factors seem to play a role in arrhythmic death in LVNC and should all be addressed in every patient. Since there is still a lack of knowledge about major phenotypical or genetic determinants of arrhythmic events, it was difficult to ignore the presence of non-sustained VT in our patient with mild LV systolic dysfunction and its potential to evolve to sustained VT. We should also not forget that many of these factors being discussed are considered to predict arrhythmic risk in other cardiomyopathies, including hypertrophic cardiomyopathy, and that LVNC shares much genetic background with other cardiomyopathies.
In summary, to the best of our knowledge, this is the first report of LVNC associated with the p.Ala335Pro mutation in the MYH7 gene. In this case, the diagnosis of LVNC was indisputable, as it was established by multiple diagnostic criteria and by echocardiography and cardiac MRI in several family members, and LVNC was associated with LV systolic dysfunction in one patient and non-sustained VT in two patients. The familial cosegregation of this genetic variant provides strong evidence of its pathogenic role in the development of LVNC. The previous finding of this mutation in hypertrophic cardiomyopathy supports the evolving concept that the same pathogenic sarcomere mutation may be associated with different cardiomyopathy phenotypes. This case also highlights the current difficulties in clinical practice regarding risk stratification and therapeutic decision-making on ICD implantation for primary prevention of sudden cardiac death in patients with LVNC.
Conflicts of interestThe authors have no conflicts of interest to declare.
