





Coronary artery disease (CAD), characterized by an atherogenic process in the coronary arteries, is one of the leading causes of death in Madeira. The GENEMACOR (GENEs in MAdeira and CORonary Disease) study sought to investigate the main risk factors – environmental and genetic – and estimate whether a genetic risk score (GRS) improves CAD prediction, discrimination and reclassification.
MethodsTraditional risk factors and 33 CAD genetic variants were considered in a case–control study with 3139 individuals (1723 patients and 1416 controls). The multivariate analysis assessed the likelihood of CAD. A multiplicative GRS (mGRS) was created, and two models (with and without mGRS) were prepared. Two areas under receiver operating characteristic curve (area under curve (AUC)) were analyzed and compared to discriminate CAD likelihood. Net reclassification improvement (NRI) and integrated discrimination index (IDI) were used to reclassify the population.
ResultsAll traditional risk factors were strong and independent predictors of CAD, with smoking being the most significant (OR 3.25; p<0.0001). LPA rs3798220 showed a higher CAD likelihood (odds ratio 1.45; p<0.0001). Individuals in the fourth mGRS quartile had an increased CAD probability of 136% (p<0.0001). A traditional risk factor-based model estimated an AUC of 0.73, rising to 0.75 after mGRS inclusion (p<0.0001), revealing a better fit. Continuous NRI better reclassified 28.1% of the population, and categorical NRI mainly improved the reclassification of the intermediate risk group.
ConclusionsCAD likelihood was influenced by traditional risk factors and genetic variants. Incorporating GRS into the traditional model improved CAD predictive capacity, discrimination and reclassification. These approaches may provide helpful diagnostic and therapeutic advances, especially in the intermediate risk group.
A doença arterial coronária (DAC), caracterizada pelo processo aterogénico nas coronárias, é das principais causas de morte na Madeira. O Genemacor (GENEs in MAdeira and CORonary Disease) visa investigar os principais fatores de risco, ambientais e genéticos, e estimar se um score de risco genético (SRG) melhora a predição, discriminação e reclassificação da DAC.
MétodosOs fatores de risco tradicionais (FRT) e 33 variantes genéticas associadas à DAC foram considerados num estudo caso-controlo com 3139 indivíduos (1723 doentes e 1416 controlos). A análise multivariada estimou a probabilidade de DAC. Foi elaborado um SRG multiplicativo (SRGm) sendo o poder discriminativo de DAC avaliado em dois modelos, um tradicional e outro com SRG incorporado. A área abaixo da curva ROC (AUC) foi investigada em ambos. Net Reclassification Improvement (NRI) e Integrated Discrimination Index (IDI) foram usados para reclassificar a população.
ResultadosTodos os FRT foram preditores para DAC, sendo o tabagismo mais significativo (OR:3,25; p<0,0001). O LPA rs3798220 apresentou maior suscetibilidade para DAC (OR:1,45; p<0,0001). Os indivíduos no quarto quartil do SRGm apresentaram 136% de probabilidade de DAC (p<0,0001). O modelo tradicional estimou uma AUC de 0,73, aumentando para 0,75 após inclusão do SRGm (p<0,0001). O NRI contínuo reclassificou melhor 28,1% da população e o categórico o grupo de risco intermédio.
ConclusõesA predição de DAC foi influenciada por FRT e variantes genéticas. O SRG incorporado no modelo tradicional, melhorou a discriminação e reclassificação de DAC. Esta abordagem pode proporcionar um diagnóstico precoce permitindo ajustes terapêuticos individualizados, especialmente no grupo de risco intermédio.
Atherosclerotic cardiovascular disease (ASCVD) has long been the first cause of death and morbidity worldwide, representing 28.7% of total deaths in the Madeira Archipelago.1,2
In response to this severe condition, the GENEMACOR (GENEs in MAdeira and CORonary disease) study was designed, early in the 2000s, to understand better the main environmental or genetic risk factors for coronary artery disease (CAD) in this archipelago. Studies based on similar aspects, including the main risk factors with an extended follow-up of all participants, have proven valuable in finding causes of heart diseases and cancer.3
Although traditional risk factors are routinely used to estimate CAD likelihood and the probability of future events, the addition of new risk factors (laboratory, clinical and genetic) can improve the performance of risk prediction models.4 Genetic information plays a prominent role in the etiology of CAD, as heritability is estimated between 40% and 60% based on family and twin studies.5,6 It is important to note that heritability results were most strongly manifested in younger individuals, indicating that genetic influence is highly significant for early-onset CAD events.
However, as the effect sizes of individual single nucleotide polymorphisms (SNP) are modest, genetic influence on risk can be quantified using a metric generally referred to as a genetic risk score (GRS) or polygenic risk score.7,8 Using four versions of the weighted multi-locus GRS to improve patient selection, requiring aggressive clinical management, recent research has shown a 1.75 fold increase in the age-adjusted CAD incidence rate in individuals with a high genetic load.9 GRS is quantifiable in early life; hence it offers the potential for early risk screening and primary prevention before other conventional risk factors become informative.10 Additional evidence is also needed to evaluate the clinical utility of polygenic risk scores in different clinical settings, such as in patients with pre-existing ASCVD.
In our country, in the early 2000s, no epidemiological follow-up studies focused on the genetic causes of CAD existed. Therefore, this paper investigates the non-genetic and genetic risk factors for CAD prediction in the GENEMACOR population. Additionally, we intend to generate a GRS, evaluating its impact on discrimination and reclassification of CAD.
MethodsStudy design and populationGENEMACOR is a case–control study with an extended prospective follow-up cohort (not presented in this paper). Consecutive coronary patients were recruited from the Cardiology Department of the Funchal Hospital Center. All data were recorded in a regional quality clinical register named MADEIRA/GESTINTERNMENT, covering more than 90.0% spectrum of patients with acute coronary syndrome (ACS) and stable angina on the Madeira Archipelago with subsequent follow-up (>99.0% coverage). After stabilization and hospital discharge, only patients in the chronic phase were considered for entry into the study (≥6 months after the acute event). The control group comprised healthy volunteers without a suggestive history of CAD, recruited through electoral rolls similar to cases in terms of age and gender.
All participants, aged between 30 and 65, were born and had been resident in Madeira Archipelago for at least two generations. This population is a genetically Caucasian Southern European sample. We analyzed stratification in our population set to recognize possible genetic admixture with principal component analysis. However, no significant genetic outliers (<5.0%) were identified.11,12
The Hospital Ethics Committee approved the study under protocol number 50/2012. All patients provided written informed consent and were advised about blood sampling for genetic and relevant clinic data. According to articles 6, 15 and 16 from the UNESCO Universal Declaration on Bioethics and Human Rights, blood samples for genetic purposes are preserved in our hospital research biobank.
Data collectionData from the clinical history comprised demographic and traditional risk factors: age, gender, smoking and alcohol habits, body mass index, diabetes, dyslipidemia, physical inactivity and arterial hypertension, as previously described.13 Clinical characteristics (e.g., heart rate, creatinine clearance and pulse wave velocity (PWV)) were also registered.13
All biochemical/laboratory analyses were carried out in the Clinical Pathology Laboratory of the Central Hospital with quality accreditation, based on the Agencia de Calidad Sanitaria de Andalucía Model (international version).
Angiographically proven CAD was considered significant if one or more coronary lesions are causing ≥70.0% stenosis in at least one major epicardial coronary artery or its primary branch, or ≥50.0% of left main coronary artery stenosis.14 The presence and severity of CAD were determined at the Hemodynamic Laboratory by two independent interventional cardiologists and, subsequently, by a research center member, whenever there was disagreement between the two.
All controls attended a medical consultation which included traditional risk factors data collection and a basal electrocardiogram. Whenever deemed necessary by the physician, other tests were required (e.g., treadmill test). Calcium scoring may also have been needed, and individuals with a score >100 Agatston units were excluded.
Genetic informationSingle nucleotide polymorphisms selectionWe included SNPs previously associated with CAD or its main risk factors from the genome-wide association studies (GWAS) or candidate gene association studies.15–19 The entering criteria comprised variants from these studies with an odds ratio (OR) for CAD ≥1 and, at the same time, with a minor allele frequency (MAF) ≥2.0%. Deviation from Hardy–Weinberg equilibrium for genotypes at the individual locus was evaluated by the Chi-squared test (p<0.002), with a multiple-comparison correction by Bonferroni for all incorporated SNPs.
In total, we considered 33 variants, which were distributed across five major physiopathological axes, according to their most consensual action pathway in coronary atherosclerosis.18
Genotype analysisA TaqMan allelic discrimination assay was used to genotype 33 genetic variants using labeled probes and primers pre-established by the supplier (Applied Biosystems). All reactions were completed on an Applied Biosystems 7300 Real-Time PCR System, utilizing the 7300 System SDS Software (Applied Biosystems, Foster City, USA). Genotypes were determined without any prior knowledge of an individual's clinical data.
Statistical analysisA comparison of baseline and biochemical data in cases and controls was performed by the Chi-squared test for categorical variables and by t Student or Mann–Whitney tests for numerical variables, as appropriate.
We collected information on the disease risk in subjects with different genotypes (for a bi-allelic polymorphism: gg, Gg, GG), performing two separate pairwise comparisons assuming a specific underlying genetic model (recessive, additive, allelic and dominant) to evaluate gene-disease associations. This method was performed by selecting the most significant OR among the genetic models for each variant.
Statistical analyses were performed using Statistical Package for the Social Sciences software version 25.0, MedCalc version 14.10.2.0 and R version 4.0.3. All p values were two-sided, statistically significant for p<0.05.
Computation of the genetic risk scoreThe GRS was composed of only 32 genetic variants, as the LPA gene variant was excluded due to its low Hardy–Weinberg equilibrium (p<0.002).
We established various models to construct the GRS using both non-weighted and weighted scores, considering each pattern of inheritance for each gene locus. This study used a multiplicative score (mGRS), obtained by multiplying the OR for each genotype.
The discriminative capacity of the mGRS was evaluated by calculating the area under the receiver operating characteristic (ROC) curve (AUC), which may be considered a global estimate of the model's predictive power. Two AUCs were performed based on two traditional risk factors multivariate models (without and with mGRS) and compared using the DeLong test. The Hosmer–Lemeshow test assessed the calibration of the models.
Coronary artery disease risk reclassificationNet reclassification improvement (NRI) was computed according to categorical NRI (cNRI) and category-free or continuous NRI (cfNRI).20,21
The contribution of mGRS, assessed using the cNRI method, evaluates the proportion of subjects moving accurately or inaccurately from one risk category to another after adding the new marker.
Participants were stratified into four predefined risk categories21,22: <25%; 25% to <50%; 50% to <75% and ≥75%, and two models (with and without mGRS) were constructed. Unlike cNRI, cfNRI does not consider pre-established risk categories but merely the direction of change (upwards and downwards) when adding the new marker.
The integrated discrimination index (IDI) measures incremental predictive value and can be viewed as the difference between average sensitivity and average 1−specificity improvement.23
ResultsOverall characteristics of the GENEMACOR populationA total of 3139 participants were evaluated, including 1723 cases (78.7% male) and 1416 controls (77.5% male). The overall characteristics of the studied population are shown in Table 1.
Overall characteristics of the study population.
| Overall characteristics | Cases(n=1723) | Controls(n=1416) | p value |
|---|---|---|---|
| Demographic | |||
| Age, years | 53±7.9 | 53±7.8 | 0.195 |
| Male sex, n (%) | 1356 (78.7) | 1097 (77.5) | 0.407 |
| Traditional risk factors | |||
| Smoking status, n (%) | 815 (47.3) | 340 (24.0) | <0.0001 |
| CAD family history, n (%) | 412 (23.9) | 186 (13.1) | <0.0001 |
| BMI, kg/m2 | 28.7±4.4 | 28.2±4.4 | 0.001 |
| Diabetes, n (%) | 582 (33.8) | 190 (13.4) | <0.0001 |
| Dyslipidemia, n (%) | 1529 (88.7) | 995 (70.3) | <0.0001 |
| Physical inactivity, n (%) | 1079 (62.6) | 612 (43.2) | <0.0001 |
| Hypertension, n (%) | 1223 (71.0) | 750 (53.0) | <0.0001 |
| SBP, mmHg | 138±20.6 | 136±17.8 | 0.046 |
| DBP, mmHg | 82±11.8 | 84±10.9 | <0.0001 |
| Alcohol >300 g/week, n (%) | 282 (16.4) | 196 (13.8) | 0.050 |
| Biochemical/laboratory risk factors | |||
| Fasting glucose, mg/dL | 105.0 (53.0–458.0) | 99.0 (71.0–393.0) | <0.0001 |
| Total cholesterol, mg/dL | 181.0 (77.0–437.0) | 203.0 (92.0–369.0) | <0.0001 |
| LDL, mg/dL | 106.2 (15.6–298.0) | 125.8 (9.6–282.6) | <0.0001 |
| HDL, mg/dL | 42.0 (18.2–115.8) | 48.0 (12.0–116.0) | <0.0001 |
| Triglycerides, mg/dL | 137.0 (31.0–2500.0) | 122.0 (30.0–1361.0) | <0.0001 |
| Apolipoprotein B, mg/dL | 93.4 (4.9–256.9) | 93.4 (3.8–212.7) | 0.023 |
| Fibrinogen, mg/dL | 386.0 (91.0–832.0) | 369.5 (127.0–1005.0) | <0.0001 |
| Homocysteine, mg/dL | 11.9 (3.7–220.2) | 11.8 (2.9–109.9) | <0.0001 |
| Homocysteine >10 mg/dL, n (%) | 1333 (77.4) | 1032 (72.9) | 0.004 |
| CRP, mg/L, n (%) | 2.7 (0.1–418.0) | 2.7 (0.1–645.0) | <0.0001 |
| Lipoprotein (a), mg/dL | 17.0 (0.5–241.0) | 14.3 (0.6–236.0) | <0.0001 |
| Lipoprotein (a) ≥30 mg/dL, n (%) | 656 (38.1) | 333 (23.5) | <0.0001 |
| Leucocytes, mg/dL | 7.1 (3.2–24.6) | 6.6 (2.1–19.7) | <0.0001 |
| Hemoglobin, mg/dL | 14.6 (8.6–18.2) | 14.7 (8.2–18.1) | <0.0001 |
| Clinical risk factors | |||
| Heart rate, beat/min | 68.4±12.6 | 72.0±11.6 | <0.0001 |
| Chronic kidney disease*, n (%) | 126 (7.3) | 50 (3.5) | <0.0001 |
| PWV, m/s | 8.7±2.4 | 8.3±1.7 | <0.0001 |
CRP: C-reactive protein; DBP: diastolic blood pressure; HDL: high-density lipoprotein; LDL: low-density lipoprotein; SBP: systolic blood pressure. Continuous variables are presented as mean±SD or median (min−max).
Statistical significance is represented by bold values.
The CAD cohort exhibits a more atherogenic profile than the control group. Specifically, CAD patients presented a higher prevalence of the main risk factors for CAD: smoking habits (47.3% vs. 24.0%; p<0.0001), CAD family history (23.9% vs. 13.1%; p<0.0001), higher body mass index (28.7 vs. 28.2; p=0.001), diabetes (33.8% vs. 13.4%; p<0.0001), dyslipidemia (88.7% vs. 70.3%; p<0.0001), physical inactivity (62.6% vs. 43.2%; p<0.0001) and arterial hypertension (71.0% vs. 53.0%; p<0.0001). The CAD cohort presented lower diastolic blood pressure values in relation to controls (p<0.0001).
All the laboratory risk factors were significantly higher in the CAD cohort (all p<0.05), except total cholesterol, LDL and HDL were higher in controls (p<0.0001). Chronic kidney disease (creatinine clearance<60 mL/min) and arterial rigidity (PWV) were significantly higher in CAD patients (p<0.0001) (Table 1).
Traditional risk factors and coronary artery disease predictionAfter the first multivariate analysis adjusted for the main classical risk factors, all were strong and independent predictors of CAD, with statistical significance (p<0.0001). “Smoking status” had the highest impact on CAD incidence, with an OR of 3.25 (95% confidence interval (CI): 2.75-3.84; p<0.0001) (Supplementary material S1), as well as after the second logistic regression, adjusted for the other confounders (OR 3.40; 95% CI: 2.83-4.09; p<0.0001). Regarding laboratory risk factors, lipoprotein (a) was the most significant CAD predictor (OR 1.85; 95% CI: 1.55-2.22; p<0.0001) (Table 2).
Traditional and new risk factors and CAD prediction.
| Variables | OR (95% CI) | p value |
|---|---|---|
| Smoking status | 3.40 (2.83–4.09) | <0.0001 |
| CAD family history | 2.07 (1.67–2.57) | <0.0001 |
| Diabetes | 2.84 (2.30–3.50) | <0.0001 |
| Dyslipidemia | 2.70 (2.18–3.35) | <0.0001 |
| Physical inactivity | 1.79 (1.51–2.11) | <0.0001 |
| Hypertension | 1.91 (1.60–2.27) | <0.0001 |
| Fibrinogen | 1.00 (1.00–1.00) | 0.002 |
| Homocysteine >10 | 1.28 (1.06–1.55) | 0.012 |
| Lipoprotein (a) ≥30 | 1.85 (1.55–2.22) | <0.0001 |
| Leucocytes | 1.07 (1.02–1.12) | 0.006 |
| Chronic kidney disease | 1.60 (1.08–2.39) | 0.020 |
CI: confidence interval. Forward Wald method by multivariate analysis. Variables that did not remain in the equation: BMI; Apolipoprotein B; CRP; PWV.
Statistical significance is represented by bold values.
In our population, two genetic variants presented minor allelic frequencies <10.0%: LPA rs3798220 (MAF=2.0%) and PPARG rs1801282 (MAF=8.7%). The association with CAD (OR) found in our population ranged from 0.86 (PON1 rs662) to 1.45 (LPA rs3798220) in the best genetic model. In these three genetic variants PON1 rs662, MTHFR rs1801131 and AGT rs699, the OR was <1 (Table 3).
Genetic variants associated with coronary artery disease in the GENEMACOR population (n=3139).
| SNP ID | Nearest gene | Alleles | CHR | OR (95% CI) | p value | MAF (%) | Putative function21 |
|---|---|---|---|---|---|---|---|
| rs599839 | PSRC1 | G>A | 1 | 1.153 (0.816–1.631)# | 0.419 | 21.3 | Lipid metabolism |
| rs2114580 | PCSK9 | A>G | 1 | 1.053 (0.914–1.213)* | 0.473 | 26.4 | Lipid metabolism |
| rs20455 | KIF6 | T>C | 6 | 1.165 (0.933–1.456)* | 0.178 | 33.0 | Lipid metabolism |
| rs3798220 | LPA | T>C | 6 | 1.445 (1.187–1.759)+ | <0.0001 | 2.0 | Lipid metabolism |
| rs964184 | ZPR1 | C>G | 11 | 1.109 (0.972–1.265)+ | 0.125 | 17.6 | Lipid metabolism |
| rs7412/rs429358a | APOEa | ɛ4 | 19 | 1.272 (1.079–1.500)# | 0.004 | 13.4 | Lipid metabolism |
| rs266729 | ADIPOQ | C>G | 3 | 1.144 (1.017–1.287)• | 0.025 | 23.5 | Diabetes/Obesity |
| rs4402960 | IGF2BP2 | G>T | 3 | 1.113 (0.874–1.419)* | 0.385 | 30.5 | Diabetes/Obesity |
| rs1801282 | PPARG | C>G | 3 | 1.154 (0.968–1.375)• | 0.110 | 8.7 | Diabetes/Obesity |
| rs1326634 | SLC30A8 | T>C | 8 | 1.064 (0.950–1.192)• | 0.281 | 25.9 | Diabetes/Obesity |
| rs7903146 | TCF7L2 | C>T | 10 | 1.011 (0.815–1.253)# | 0.924 | 35.2 | Diabetes/Obesity |
| rs1376251 | TAS2R50 | G>A | 12 | 1.184 (0.623–2.250)* | 0.606 | 11.9 | Diabetes/Obesity |
| rs8050136 | FTO | C>A | 16 | 1.180 (1.020–1.366)# | 0.026 | 39.7 | Diabetes/Obesity |
| rs17782313 | MC4R | T>C | 18 | 1.369 (0.987–1.897)* | 0.059 | 21.7 | Diabetes/Obesity |
| rs1884613 | HNF4A | C>G | 20 | 1.129 (0.968–1.317)* | 0.123 | 16.2 | Diabetes/Obesity |
| rs699 | AGT | T>C | 1 | 0.958 (0.825–1.112)# | 0.573 | 42.6 | Hypertension |
| rs5186 | AGT1R | A>C | 3 | 1.075 (0.957–1.207)+ | 0.225 | 24.9 | Hypertension |
| rs4340 | ACE | I>D | 17 | 1.141 (0.987–1.320)* | 0.074 | 38.3 | Hypertension |
| rs1801131 | MTHFR | A>C | 1 | 0.939 (0.815–1.080)* | 0.376 | 28.2 | Oxidation |
| rs1801133 | MTHFR | C>T | 1 | 1.206 (1.047–1.389)# | 0.010 | 33.5 | Oxidation |
| rs6922269 | MTHFD1L | G>A | 6 | 1.063 (0.811–1.392)# | 0.660 | 27.3 | Oxidation |
| rs705379 | PON1 | C>T | 7 | 1.062 (0.898–1.256)# | 0.483 | 46.9 | Oxidation |
| rs662 | PON1 | A>G | 7 | 0.861 (0.678–1.094)# | 0.219 | 29.9 | Oxidation |
| rs854560 | PON1 | T>A | 7 | 1.130 (1.021–1.251)• | 0.018 | 40.7 | Oxidation |
| rs17465637 | MIA3 | A>C | 1 | 1.063 (0.953–1.186)• | 0.272 | 28.9 | Cellular |
| rs618675 | GJA4 | T>C | 1 | 1.046 (0.923–1.186)* | 0.484 | 19.6 | Cellular |
| rs12190287 | TCF21 | G>C | 6 | 1.260 (1.094–1.452)+ | 0.001 | 32.9 | Cellular |
| rs1332844 | PHACTR1 | C>T | 6 | 1.129 (1.021–1.248)* | 0.018 | 44.5 | Cellular |
| rs11556924 | ZC3HC1 | T>C | 7 | 1.137 (1.023–1.263)+ | 0.017 | 34.2 | Cellular |
| rs1333049 | CDKN2B-AS1 | G>C | 9 | 1.143 (1.035–1.263)• | 0.009 | 45.6 | Cellular |
| rs4977574 | CDKN2B-AS1 | A>G | 9 | 1.161 (1.050–1.284)• | 0.004 | 41.9 | Cellular |
| rs17228212 | SMAD3 | T>C | 15 | 1.110 (0.830–1.483)* | 0.483 | 25.3 | Cellular |
| rs3825807 | ADAMTS7 | A>G | 15 | 1.077 (0.974–1.192)• | 0.149 | 41.4 | Cellular |
Chr: chromosome. The table shows susceptible loci for CAD, ORs and p values for the lead SNP within each locus. ORs are given for additive, recessive, allelic or dominant models, according to the most significant. The potential mechanism of action is based on what is already known about the function of the nearby genes.
Statistical significance is represented by bold values.
In the bivariate analysis, gene by gene, according to the main genetic inheritance models, we found 11 statistically significant variants associated with CAD, namely: TCF21 rs12190287 and CDKN2B-AS1 rs4977574 remained significant in all previously described genetic models; LPA rs3798220, APOE rs7412/rs429358, ADIPOQ rs266729, FTO rs8050136, MTHFR rs1801133, CDKN2B-AS1 rs1333049, PON1 rs854560, PHACTR1 rs1332844 and ZC3HC1 rs11556924 maintained significance in only three genetic models (Supplementary material S2).
Genetic variants and coronary artery disease predictionOnly nine genetic variants remained independent CAD predictors after adjusting for all the significant genetic variants from the bivariate analysis (Table 3). LPA rs3798220 was the most significant variant for CAD with an OR of 1.43 (95% CI: 1.17–1.74; p<0.0001) (Table 4).
Genetic variants independently associated with coronary artery disease.
| Variables | OR (95% CI) | p value |
|---|---|---|
| LPA rs3798220 | 1.43 (1.17–1.74) | <0.0001 |
| APOE rs7412/rs429358 | 1.21 (1.05–1.41) | 0.010 |
| TCF21 rs12190287 | 1.20 (1.08–1.34) | 0.001 |
| CDKN2B-AS1 rs4977574 | 1.16 (1.05–1.28) | 0.004 |
| ADIPOQ rs266729 | 1.15 (1.02–1.29) | 0.022 |
| PON1 rs854560 | 1.13 (1.02–1.25) | 0.023 |
| PHACTR1 rs1332844 | 1.12 (1.01–1.24) | 0.028 |
| MTHFR rs1801133 | 1.12 (1.01–1.25) | 0.038 |
| ZC3HC1 rs11556924 | 1.12 (1.00–1.24) | 0.044 |
Forward Wald method. The variants that did not remain in the equation were: CDKN2B-AS1 rs1333049 and FTO rs8050136.
Statistical significance is represented by bold values.
Eight genetic variants maintained CAD significance after adjusting for the significant traditional risk factors from the previous analysis (Figure 1). LPA was the most important genetic risk factor with an OR of 1.51 (95% CI: 1.21–1.87; p<0.0001), followed by APOE (OR 1.25; 95% CI: 1.06–1.47; p=0.007) and CDKN2B-AS1 rs4977574 (OR 1.19; 95% CI: 1.07–1.32; p=0.002) (Figure 1).
Genetic risk score and coronary artery disease prediction
The mGRS was constructed and presented a mean of 1.16±0.77 in the CAD cohort and 0.96±0.67 in controls. Afterwards, it was subdivided into quartiles, and the results showed that as the mGRS quartile rises, there is an increase of cases and decrease among controls and, subsequently, an increase in CAD prediction (p<0.0001) (Figure 2).
Coronary artery disease predictive power of the mGRS added to traditional risk factors
The inclusion of GRS to traditional risk factors increased the discriminative power of CAD (AUC=0.75) when compared to the traditional model (AUC=0.73), with statistical significance (p<0.0001). The Hosmer-Lemeshow test presented a good calibration for the two models, with and without mGRS (p=0.619 and p=0.996, respectively), and was for this reason, well-fitted to predict CAD in our population.
The mGRS predicts CAD likelihood more accurately than dyslipidemia and hypertension, presenting an AUC of 0.60 (identical to diabetes). “Smoking status” was the traditional risk factor with better capacity to predict CAD (AUC=0.62) (Figure 3).
Coronary artery disease risk reclassificationCategorical net reclassification improvement.")
The mGRS improved risk reclassification, particularly in the intermediate-risk categories (25% to <50% and 50% to <75%). In our study, the model with mGRS was more accurate in 382 cases (22.2%) and less accurate in 200 (11.6%), thus accounting for a net gain reclassification of 10.6% of subjects with CAD. In the control group, the percentages were 15.5% and 15.4%, respectively (net gain reclassification of 0.1%), which resulted in an improved reclassification of 10.7% of the total population (Table 5 and Figure 4).
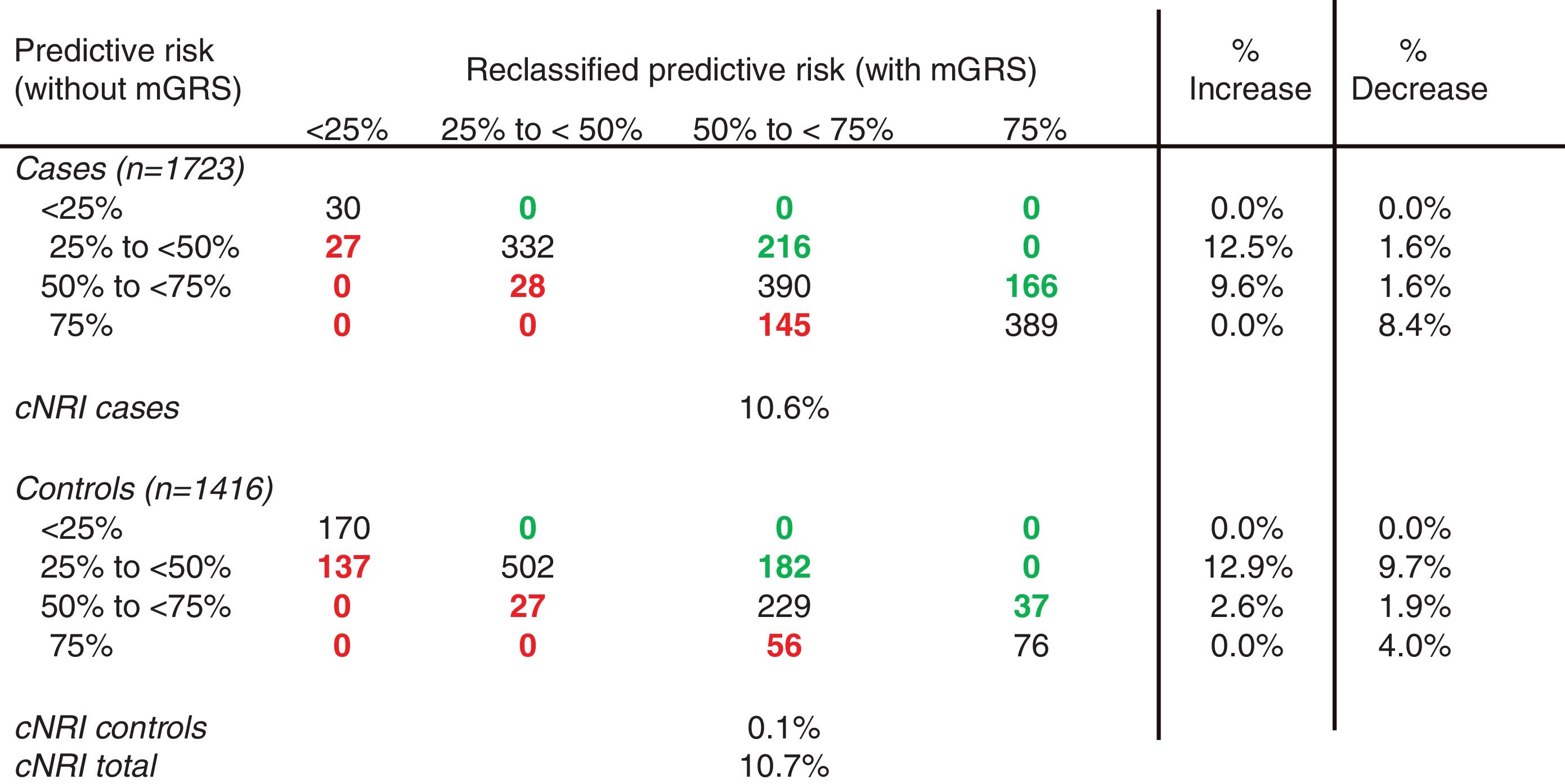
 into four risk categories. Coronary patients are better reclassified for intermediate/high and higher-risk categories, and controls are better reclassified for low and low/intermediate-risk categories.")
In this study, the proportion of CAD patients whose calculated risk increased, minus the percentage whose risk decreased was 12.7%. In contrast, the proportion of individuals without CAD whose risk reduced, minus the ratio whose risk increased was 15.4%, giving a total cfNRI of 28.1%. The inclusion of mGRS in the traditional risk factors provided an IDI of 0.019 (95% CI: 0.014–0.024) (Table 6). It indicates there is a statistical difference between the two models (p<0.0001).
DiscussionIn the current work, some relevant findings from the GENEMACOR population were described: first, the impact of the traditional risk factors on CAD predisposition and second, the effect of the genetic component in CAD likelihood. Finally, we demonstrated that adding genetic variants, quantified through a GRS, to traditional risk factors improved CAD predictive capacity, discrimination and reclassification.
In this study, the prevalence of conventional risk factors ranged from 33.8% in diabetic patients to 88.7% in patients with residual dyslipidemia, despite treatment in the context of secondary prevention. A lower atherogenic risk profile was found for the control population in a primary prevention setting. Smoking is very prevalent in the CAD population. Almost half of these were smokers, representing a critical risk factor, with a 3.40-fold increased likelihood for CAD, even after multivariate adjustment for traditional, biochemical and clinical risk factors. Similarly, previous studies have shown a two- to four-fold increased risk of CAD, and an elevated mortality rate, including sudden death.24 One cardiovascular prevention objective according to the 2021 European Society of Cardiology (ESC) guidelines would be to achieve a smoke-free Europe by 2030.
The demonstration of the association between traditional risk factors and CAD is relevant, although not innovative. However, the statistical appearance of traditional risk factors as predictive of CAD validates the study protocol and supports the results of genetic analysis, the main focus of the present paper.
Evidence shows that CAD mortality strongly correlates with environmental and lifestyle variances. Nevertheless, individuals with identical cardiovascular risk factors such as smoking, serum cholesterol and blood pressure levels can present different CAD mortality and cardiovascular event rate. Specific genetic characteristics may explain those differences in regions with the same level of conventional risk factors. We investigated 33 genetic variants in the GENEMACOR population, but we only discuss eight variants independently associated with CAD in the multivariate analysis with traditional risk factors. All were in Hardy–Weinberg equilibrium, apart from the LPA variant. However, this rare variant rs3798220 T>C had the most potent CAD association effect. It is a highly polymorphic and informative copy number variation with 95% heterozygosity in most European Caucasian populations.25–27 In our population, this variant did not present CC homozygosity. Scarce variants are underrepresented in the human genome, attributed to purifying selection: alleles with a more significant effect are more expected to be harmful and less likely to increase in frequency in the variants that promote the disease.
The PROCARDIS CONSORTIO25 showed that patients who carried at least one risk allele of this SNP had a 1.51-fold elevated probability for myocardial infarction and subjects carrying two risk alleles had a 2.57-fold increase in CAD risk. After adjustment for confoundable variables, it remained significant with an OR of 1.51. These findings are in accordance with a recent study that highlighted the LPA rs3798220 as strongly associated with Lp(a) concentrations and CAD.28
The LPA gene codifies the Lp(a) protein, the lipoprotein with the most potent genetic control. In the present study, 38.1% of our patient population and 23.5% of the controls had Lp(a) levels >30 mg/dl, representing a decisive risk factor for CAD with an OR of 1.85. Recent evidence has proved that high levels of this atherothrombotic risk factor can become a therapeutic target.29 Future developments in antisense oligonucleotides to lower Lp(a) can assess whether this reduction decreases the likelihood of CAD.
The APOE variant, composed of two mutations (rs7412 and rs429358), originates three main protein variants, Apo E2, E3 and E4. The corresponding genotypes, ɛ2/2, ɛ2/3, ɛ2/4, ɛ3/3, ɛ3/4 and ɛ4/4, interfere with lipoprotein metabolism. Recent studies have shown that carriers of the ɛ3/ɛ4 and ɛ4/ɛ4 genotypes are associated with higher serum concentrations of triglycerides, total cholesterol, LDL-C, lower HDL-C and a higher predisposition to CAD.30,31 Likewise, APOE increased CAD likelihood by 24.8%, in the present study, after adjusting for traditional risk factors and all significant GENEMACOR variants. The CDKN2B-ASI rs4977574 (ANRIL) in the 9p21 genomic region is a non-coding RNA adjacent to cell cycle regulating genes, CDKN2A and CDKN2B. Although there is no consensus, ANRIL may increase the expression of these genes through the proliferation of vascular smooth muscle cells in atherosclerotic plaques.32 In our study, the probability for CAD was 1.19 after adjustment for traditional and genetic risk factors.
The TCF21 variant rs12190287 is a transcription factor whose pathophysiological function is not consensual and may involve processes related to vascular gene expression and smooth muscle cell biology.33 It was recently associated with events and may represent a new attractive target to reduce cardiovascular risk.34,35 Our GENEMACOR population presents a CAD probability of 1.17, consistent with other European studies.
The following two SNPs, belonging to the cellular pathophysiologic axis, are also involved in the cell cycle regulation: ZC3HC1 rs11556924 and PHACTR1 rs1332844. ZC3HC1 codifies a complex protein (nuclear Interaction Partner ALK – NIPA) that presents lower regulatory NIPA phosphorylation, which originates elevated protein activity with higher mitotic rate and CAD risk.36PHACTR1 rs1332844 encodes actin regulatory protein phosphatase, regulating essential cell functions such as cell migration and survival.37 Both SNPs presented similar CAD likelihood to the TCF21 variant (OR=1.16).
The last significant SNPs, PON1 rs85456038 and MTHFR rs1801133,39 belong to the oxidative axis presenting an OR of 1.15 and 1.13, respectively. MTHFR is widely regarded as the leading cause of hyperhomocysteinemia (HHcy). This SNP may affect DNA methylation and modify the lipid metabolism inducing endothelial dysfunction and atherogenic process. Among the GENEMACOR CAD patients, plasma Hcy levels were significantly higher than controls, presenting an OR for CAD of 1.28.
As the effect sizes of individual SNPs were modest, a GRS containing several SNPs was considered. The total burden of genetic risk for CAD is proportional to the number of inherited variants, and the risk can be combined and quantified into a single number capable of predicting CAD. The 2021 ESC guidelines on CV disease prevention have shown some potential of the polygenic GRS to improve ASCVD risk prediction for primary and secondary prevention. The present study found that using a polygenic risk score added to traditional risk factors significantly improved CAD risk prediction and reclassification, specifically in individuals belonging to the intermediate risk category. The reclassification to high or low risk may change the therapeutic approach to the patient40 allowing for individualized therapeutic adjustment in preventing future CAD cases or delaying its evolution throughout life.
Strengths and limitationsThe main strength of this study arises from the fact that it was carried out in Madeira with participants born and living in a genetically isolated area. It consists of a homogeneous population due to its isolation for many years, with no genetic admixtures.12,41,42 Most studies with GRS include heterogeneous populations. This methodology is subject to criticism for comparing different populations, thus influencing genetic research.
Another strength is the proven clinical utility of GRS performed through a set of significant SNPs that improved CAD risk reclassification. The potential value of incorporating genetic information besides the phenotype with traditional risk factors in common diseases may allow better decision-making on drug therapy, especially in lipid-lowering drugs.
One limitation concerns the restricted number and type of SNPs included in our GRS since more specific and powerful SNPs could have been incorporated, as well as rare forms that could still increase their predictive capacity and accuracy in overall prediction and reclassification.
Another limitation is that as it is a case-control study, by definition, only survival cases can be included in the study. Therefore, all deaths before reaching the hospital or in the acute disease phase were not included. All patients entered this study in the chronic phase.
This study only included individuals from Madeira aged between 30 and 65 years. Therefore results may not be extrapolated to other populations or age-groups, including >65 years which represents a significant proportion of CAD patients. However, the genetic influence is most important in younger individuals by allowing early preventive strategies before other conventional risk factors become informative.
Furthermore, the definition of CAD was evaluated by local site interpretation of coronary angiography and not by a central core laboratory. The blinded assessment of clinical information may avoid interpretation bias.
ConclusionSusceptibility to cardiovascular disease in the GENEMACOR population was influenced by traditional risk factors and genetic variants. Combining traditional and genetic information incorporated in a polygenic risk score improved coronary disease prediction, discrimination and reclassification capacity. These approaches may provide clinically helpful advances in personalized coronary disease control over the next decade. The time for tailored medicine has, in fact, arrived.
Conflicts of interestThe authors have no conflicts of interest to declare.
The authors are very grateful to Mrs Elsa Segura for all the administrative support and to Mrs Graça Correia and Mrs Paula Nascimento from the hospital's library for efficient scientific research. Special gratitude goes to Dr Tiago Mendonça for the helpful language review.
The following are the supplementary data to this article:

