




Abnormal arrangement of thoracoabdominal organs, situs ambiguous, is also known as heterotaxy syndrome (HTX). It has been frequently linked with congenital heart diseases (CHD), which are commonly reported as atrioventricular septal defects (AVSD), atrial septal defects (ASD), ventricle septal defects (VSD), transposition of the great artery, and pulmonary stenosis or atresia. Two HTX categories are right atrial isomerism (RAI) and left atrial isomerism (LAI), which are distinguished by the organ's sidedness as well as their complexity. The etiology of the syndrome is being studied widely, where recent studies are more focused on the effect of gene variants present in affected individuals. DNAH11, DNAH5, ZIC3, NODAL, and LEFTY are among the genes studied and associated with HTX. DNAH11 and DNAH5 are associated with ciliary function while ZIC3, NODAL and LEFTY are associated with signaling pathways. As multiple genes are involved, HTX has been reported to have an autosomal dominant, autosomal recessive or X-linked inheritance patterns, depending on the causative genes in the individual. This review aims to summarize several previously reported HTX gene variants, inheritance patterns, as well as the cardiac and extracardiac clinical manifestations.
O arranjo anormal dos órgãos toracoabdominais, situs ambiguus, também é conhecido como síndrome de heterotaxia (HTX). HTX é frequentemente associada a doenças cardíacas congénitas (DCC) que podem ser descritas como defeitos do septo atrioventricular (DSAV), defeitos do septo atrial (DSA), defeitos no septo ventricular (DSV), transposição das grandes artérias (TGA) e estenose pulmonar ou atresia. Duas categorias do HTX são o isomerismo atrial direito (IAD) e isomerismo atrial esquerdo (IAE), que se distinguem pela orientação lateral do órgão, assim como pela sua complexidade. A etiologia desta síndrome está a ser amplamente investigada, onde os estudos mais recentes têm-se focado no efeito de variantes genéticas em indivíduos afetados. DNAH11, DNAH5, ZIC3, NODAL e LEFTY estão entre os genes mais estudados em relação à HTX. DNAH11 e DNAH5 estão associados à função ciliar, enquanto ZIC3, NODAL e LEFTY estão relacionados com vias de sinalização. Uma vez que vários genes estão implicados, a HTX pode apresentar herança autossómica dominante, autossómica recessiva ou ligada ao X, dependendo dos genes causativos. Esta revisão tem como objetivo resumir as diversas variantes genéticas anteriormente descritas e associadas à HTX, bem como padrões de herança e manifestações clínicas cardíacas e extracardíacas.
Heterotaxy syndrome (HTX) is an abnormality where internal thoracoabdominal organs are arranged abnormally along the left–right axis of the body, where they diverge from the regular arrangement (situs solitus) and the complete mirror of it (situs inversus)1–3 as depicted in Figure 1. The divergence from the customarily accepted arrangement of left–right patterning is mainly observed in the arrangement of the appendages of the atria and the spleen.2 Any arrangement that is not categorized as situs solitus or situs inversus may be described as situs ambiguous, and this categorization is mainly used for HTX.4
 Situs solitus is the normal arrangement of the thoracoabdominal organs. (B) Situs inversus is the complete mirror of situs solitus. (C) Right atrial isomerism with bilateral trilobed lungs, bilateral right atrial appendages, central liver, and asplenia. (D) Left atrial isomerism with bilateral bilobed lungs, bilateral left atrial appendages, central liver, and polysplenia. L: left; LAA: left atrial appendage; R: right; RAA: right atrial appendage.")
General arrangement of the thoracoabdominal organs in normal individuals and individuals with situs abnormalities, including HTX. (A) Situs solitus is the normal arrangement of the thoracoabdominal organs. (B) Situs inversus is the complete mirror of situs solitus. (C) Right atrial isomerism with bilateral trilobed lungs, bilateral right atrial appendages, central liver, and asplenia. (D) Left atrial isomerism with bilateral bilobed lungs, bilateral left atrial appendages, central liver, and polysplenia. L: left; LAA: left atrial appendage; R: right; RAA: right atrial appendage.
Congenital heart disease (CHD) is a prevalent birth anomaly, occurring in approximately 8–12 out of every 1000 live births globally.5 HTX is an uncommon birth disorder that affects around 1 out of every 10000 live births, with the majority of them complicated by complex CHD.6 It is noteworthy that there is a higher occurrence of heterotaxy syndrome among individuals with CHD. Research studies have indicated that approximately 5–10% of CHD patients exhibit heterotaxy syndrome.7 The prognostication of the disease, primarily in children with right isomerism with complex congenital defects, has been poor despite the advancement in medical and surgical treatments.8,9
Currently, the diagnosis is made mainly by observing the spleen and the heart morphology prenatally with ultrasound, which is challenging to validate; hence, any observed abnormalities are compared with the abnormalities found postnatally.1 The integration of echocardiography and genetic diagnosis such as whole exome sequencing has been proposed to produce better diagnoses.10
Research on the genetic factors contributing to the association between HTX and CHD is ongoing, and the exact mechanisms are not fully understood. However, there have been some studies indicating potential genetic links between these conditions.6,11–14 Studies have shown that variants in genes causing primary ciliary dyskinesia (PCD) may be associated with the development of HTX with or without CHD.6 For example, variants in the DNAH11 gene have been linked to the development of HTX and CHD.6 Additionally, the presence of copy number variations has also been identified as a genetic basis for HTX and various CHD.13 As further research continues, more genes associated with situs ambiguous are being identified, highlighting the genetic complexity of these conditions.14
Multiple genes have been associated with HTX, with various modes of inheritance, including autosomal recessive, autosomal dominant, and X-linked, have been observed.15 Hence, the aim of this review is to summarize the genes with previously reported variants associated with HTX (DNAH11, DNAH5, ZIC3, NODAL, and LEFTY), their inheritance patterns, and clinical manifestations. These genes are a part of the suggested etiology of HTX, ciliary function as well as signaling pathways.
Heterotaxy syndromeTwo classes of isomerism are used to categorize HTX further. Isomerism is described as the arrangement where organ structures that are supposedly situated on the right or left side of the body are seen on both sides instead.16 The isomeric arrangements may only be seen in certain organs of the individual, and these abnormal arrangement combinations are still considered as isomerism.2
Isomerism in HTX can be divided into two classifications: left atrial isomerism (LAI) and right atrial isomerism (RAI) (Figure 1). However, some patients may manifest both features. When the thoracic or abdominal cavity organs are present with left-sided morphology, it is known as LAI, where it is manifested mainly by left appendages on both sides of the atria, as well as polysplenia, which were reported in more than half of the cases.4 The bilaterally left appendages are described as long and tube-like narrowing at the opening, while the lungs are bilobed with polysplenia.8 In contrast, RAI is right sided with broad-based and triangular shape appendages, trilobed lungs, and absence of the spleen.4
Several retrospective studies focusing on multiple populations have reported more LAI cases in Western countries, while RAI prevalence is greater in Asians.17–20 Two studies which included Germany found more than 50% of the patients reported with LAI.19,20 Another study conducted in the United Kingdom reported almost two times more LAI cases compared to RAI.18 In contrast, in Asian populations, the frequency of RAI was higher than LAI, where the LAI occurrence ratio among HTX patients in Korea, India, Taiwan, and Singapore were all above 0.53.17,21–23 As a result, it is thought future research should focus on the heterogeneity of different populations.17 In addition, HTX with CHD recurrence risk was reported to be as high as 64% in one study.24
Heterotaxy syndrome can have a significant impact on patient outcomes, particularly due to associated complex congenital heart defects and multiorgan dysfunction.25 It can impact various non-cardiac systems, including the immunologic, respiratory, and genitourinary systems, which can affect patient outcomes.26 Morbidity and mortality in patients with HTX remain high despite recent advances in surgical management.8 A study reported by Serraf et al. (2010) states that overall mortality was 20.8%, with 7.2% after the first surgery, 6.6% after the second, and 11.5% after the third.8 Long-term outcomes have not improved since the early 1990s.27 The complexity of underlying CHD in RAI is greater than LAI as the structural defects are more complicated; hence a greater infant mortality was reported.28 Prognostication of a 5-year survival from RAI is worse compared to LAI, where they fall in ranges of 30–74% for RAI and 65–84% for LAI.8
Patients with HTX need a comprehensive management approach; each child will have different needs depending on the nature and severity of the organ involvement. The management and treatment of heterotaxy patients have advanced such as the Ladd procedure for the management of malrotation in children with heterotaxy and CHD, but the prognosis remains unsatisfactory due to the association with complicated CHDs.29 Surgical outcomes in heterotaxy patients have improved in recent years, with low mortality rates after the Fontan operation.30 Proper counseling for the family about the course of the disease, treatment modalities, and outcomes is essential.31 Surgical management of heterotaxy syndrome presents unique challenges, and certain anatomic subsets may require novel strategies.32
Heterotaxy syndrome and congenital heart diseaseA spectrum of congenital heart defects had been seen in HTX with distinctive features of systemic and pulmonary venous anomalies, inlet abnormalities and conotruncal defects.33 Most patients with HTX are diagnosed with complex CHD.34 Phenotypic cardiac defect manifestations due to disruption of the left–right asymmetry are complex and extensive.15,35 Complete atrioventricular septal defects (AVSD), atrial septal defects (ASD), ventricle septal defects (VSD), transposition of the great artery, and pulmonary stenosis or atresia are the primary cardiac abnormalities related with RAI.21 On the other hand, LAI patients are more commonly associated with ventricular septal defect, atrial septal defect, and left-sided obstructive lesions.36 LAI patients also have a higher incidence of sinus node dysfunction or heart block compared to RAI patients.37 The occurrence of arrhythmias is more common in LAI patients, with more than half of them experiencing arrhythmias by the age of three.38 The anatomical variants of cardiac structures in heterotaxy patients include abnormal pulmonary veins, low pulmonary blood flow, abnormal relationship of great arteries, and atrioventricular canal defect.39
A study by Ware et al. (2004) on 93 patients reported 28 RAI patients, from among whom 29% showed pulmonary atresia, 46% with pulmonary stenosis, 68% with complete atrioventricular canal, 50% with transposition of the great arteries, and 36% with total anomalous pulmonary venous return, while the percentage of these cardiac defects were lower in LAI at 11%, 33%, 44%, 33%, and 11%, respectively.40 The interrupted inferior vena cava with azygous venous continuation is a constant sign in LAI patients.17 The percentage of levocardia is higher in LAI (89%), while dextrocardia is higher in RAI (46%).21,40 Cardiac abnormalities linked to RAI and LAI are quite similar; however, complex cardiac malformation was shown to be less common in LAI than in RAI.17,21 Besides, there are overlapping of cardiac defects associated with RAI and LAI reported such as atrioventricular septal defects, double outlet right ventricle, transposition of the great arteries and pulmonary stenosis.21
Heterotaxy syndrome is complex and diverse; for example, a study reported two fetuses from the same mother were found to have the same variants but manifested different types of isomerism and cardiac lesions.41 The occurrence of different abnormalities may be higher in LAI but lower in RAI and vice versa. Ringle et al. (2021) reported different prevalence of cardiac morphological anomalies such as pulmonary atresia, ventriculoarterial connection, arterial trunks positioning, atypical distribution of coronary artery and single coronary artery, between LAI and RAI.42 In a study by Baban et al. (2018), it was found that 81 patients were classified as RAI and 55 patients as LAI. The long-term survival and freedom from heart transplant were higher in LAI patients compared to RAI patients, reaching 87.8% at 40 years for LAI and 69.8% for RAI.43
Congenital heart disease has implications on the outcomes of heterotaxy patients. Studies have shown that heterotaxy patients with CHD have increased postoperative mortality and respiratory complications.44 These patients have higher rates of tracheostomies, extracorporeal membrane oxygenation support, and prolonged ventilatory courses.45 Heterotaxy-CHD patients with respiratory ciliary dysfunction (CD) may have even worse postsurgical outcomes, including increased respiratory complications and the need for tracheostomy.38 Lim et al. (2005) reported that most left isomerism (LAI) cases required no intervention or underwent successful biventricular cardiac surgery, unlike right isomerism (RAI) cases. The study also found that patient age at initial surgery was an independent risk factor for mortality.46
The treatment and management strategy for heterotaxy-associated congenital heart disease patients is complex and varies depending on the specific cardiac malformations present. Surgical management is often necessary, and the choice of procedure depends on the anatomical abnormalities, including systemic and pulmonary venous connections. In cases of significant ventricular hypoplasia or extreme complexity, single ventricle palliation followed by the Fontan operation is commonly performed.29 HTX remains a challenging condition with high morbidity and mortality, particularly in patients with total anomalous pulmonary vein return and right ventricular outflow tract obstruction.8 Despite significant advancements in medical and surgical treatments for heterotaxy syndrome, the long-term prognosis for patients is still unsatisfactory.
The overall short-to-mid-term prognosis of heterotaxy patients is not satisfactory, however, multiple approaches including basic and clinical science are necessary to improve the prognosis and quality of life of heterotaxy patients. Systematic follow-up of the patients will improve their prognosis. Therefore, it can be inferred that the quality of life of heterotaxy patients can be affected by the complications associated with the disease, but it can be improved through better clinical management, tailor-made surgical operations, and systematic follow-up of the patients.30 Overall, a multidisciplinary approach involving pediatric and adult congenital heart disease specialists is crucial for the successful treatment and management of heterotaxy-associated congenital heart disease patients.
Suggested etiologyThe etiology of HTX remains indefinite; however, effects from gene variants have been suggested, and genes related to ciliary function were studied the most.47 Disruption of the signaling pathway, mainly the Nodal signaling pathway, is another suggested etiology of HTX.15ZIC3 gene involvement in the Hedgehog signaling pathway and the canonical WNT signaling pathway has also been inferred.48 Environmental factors that might influence the etiology of heterotaxy include teratogenic exposures, especially maternal diabetes.49 Heterotaxy syndrome arises from the failure of intra-abdominal and intra-thoracic structures to rotate appropriately during embryologic development, which may occur due to a series of mutations in genes regulating the development of normal left–right axis determination.50
Ciliary functionGenerally, human cilia are classified into two types: non-motile cilia (also known as primary cilia) and motile cilia. The critical function of primary cilia involves the signaling pathway and the regulation of the cellular environment, while respiratory tract mucous clearing, transport, and movement of cerebrospinal fluid, as well as gametocytes, are performed by the motile cilia.51 Dextrorotatory of the cilia produces a one-way nodal flow toward the left; hence transmission of determinant molecules is then toward the lateral plate mesoderm on the left, which produces left–right patterning asymmetry of the organs.9 Cilia generate leftward fluid flows in an embryonic “left–right organizer,” which directs asymmetric activation of the Nodal signaling pathway.52 This pathway guides the asymmetric morphogenesis of developing organs, including the heart.53 Studies in mice have shown that cilia transduced Hedgehog signaling coordinates left–right patterning with heart looping and differentiation of the heart tube.54
Studies on PCD or cilia dysfunction in HTX suggested that the genes involved in ciliary function play essential roles in determining the left–right patterning.6 Examples of genes involved in ciliary structure and function are DNAH11, DNAH5, DNAH7, and DNAI1.55 At the embryonic node, abnormalities in either motile or non-motile cilia may alter the left nodal flow, leading to HTX.51 About 12.1% of patients diagnosed with PCD and about 8.9% of patients without PCD were associated with HTX.56 A recent study by Liu et al. (2019) mentioned that roughly half of the patients with PCD were reported to have HTX associated with CHD.6 HTX and CHD are caused by multiple genes while PCD is a monogenic recessive illness with genetic heterogeneity.6,47 The connection between HTX, CHD and PCD needs to be investigated further, considering the effect of ciliary function on HTX and CHD from past studies.
Signaling pathwaysHeterotaxy syndrome arises from intricate dysregulations in multiple signaling pathways during embryonic development. The Nodal, Hedgehog, and canonical WNT pathways play pivotal roles in establishing left–right asymmetry.57–59
The nodal signaling pathway is a significant component in left–right patterning determination during embryo development.60 In the Nodal signaling pathway, information is transmitted by type I and II receptors and EGF-CFC co-receptors, and the consequences of the receptor activation are SMAD2 and SMAD3 phosphorylation (Figure 2(A)).60,61 This enables SMAD2/SMAD3 binding to SMAD4, causing interactions with other transcription factors and target genes regulations in the nucleus. The heterodimeric SMAD complex may interact with DNA binding proteins (e.g. FOXH1 and MIXER) or simply bind to DNA.62 This process involves both extracellular and intracellular components.61 Proteins from the Nodal signaling pathway i.e., NODAL, LEFTY and PITX2 are expressed asymmetrically on the left side while LEFTY1 is expressed on the midline of the lateral plate mesoderm (LPM).15 The Nodal signaling pathway is dose sensitive, where any abnormality in signal transduction will affect Nodal expression, risking abnormal patterning and lead to HTX.62 Damaged Activin/Nodal-Smad signaling and reduction in signal transduction were observed in functional analysis of NODAL variants, supporting the dose sensitive hypothesis of the pathway.60,62 In an animal study, NODAL was found to have an important function in primitive streak and embryonic mesoderm development.63 To maintain balanced expression of Nodal, antagonists which are the Lefty proteins, act as negative feedback toward the Nodal expression.15,60 In a targeted study on mice, all four Lefty1 exons were removed using targeting vector, which resulted in abnormality of left–right patterning as Nodal was expressed bilaterally in the LPM.64 This supports the postulated mechanism of either the absence or bilateral expression of NODAL in NODAL signaling that causes HTX.62,64
 Nodal signaling pathway. NODAL binds to its receptors (type 1, ActRII and type 2, ALK4) and co-receptor (EGF-CFC). SMAD2/3 are phosphorylated and bind with SMAD4, forming a complex. The SMAD complex enters the nucleus to regulate target genes transcription either by binding to the DNA binding protein or directly binds to the DNA. To keep the balance of NODAL expression, LEFTY acts as the negative feedback protein where it either binds to NODAL of the co-receptor when NODAL is expressed in abundance. Repression of the target genes may lead to absence or bilateral expression of NODAL leading to laterality defects and HTX. (B) Hedgehog signaling pathway. SMO inhibition by PTCH is lifted when Hh ligands bind to PTCH. This allows downstream cascade as SuFu stops its binding to GLI and GLI is able to enter the nucleus for target gene transcription. Binding of SuFu to GLI is a negative regulation of the pathway as the binding produces GLI-repressor. Presence of ZIC3 will inhibit the production of GLI-repressor and affect the gene transcription process in the nucleus. Repression of the IFT genes will cause disruption of the ciliary structure and function which also will cause laterality defects and HTX. (C) Canonical WNT signaling pathway. WNT binds to its receptor, FZD and co-receptor, LRP5/6. The binding induces dissociation of a complex consisting of APC, AXIN/DVL/GSK3/CK1/β-catenin. The result of the dissociation is the release of β-catenin which is then able to enter the nucleus to regulate target genes transcription by interacting with TCF. ZIC3 binding to TCF has the ability to repress β-catenin, hence affecting the WNT signaling pathway. It is suggested that β-catenin repression will give effect of suppressing the WNT pathway. Another suggested effect is reduction or accumulation of β-catenin. Both lead to failure in forming the dorsal axis, disrupting organ arrangements and cause HTX.")
Three signaling pathways suggested to be involved in causing HTX. (A) Nodal signaling pathway. NODAL binds to its receptors (type 1, ActRII and type 2, ALK4) and co-receptor (EGF-CFC). SMAD2/3 are phosphorylated and bind with SMAD4, forming a complex. The SMAD complex enters the nucleus to regulate target genes transcription either by binding to the DNA binding protein or directly binds to the DNA. To keep the balance of NODAL expression, LEFTY acts as the negative feedback protein where it either binds to NODAL of the co-receptor when NODAL is expressed in abundance. Repression of the target genes may lead to absence or bilateral expression of NODAL leading to laterality defects and HTX. (B) Hedgehog signaling pathway. SMO inhibition by PTCH is lifted when Hh ligands bind to PTCH. This allows downstream cascade as SuFu stops its binding to GLI and GLI is able to enter the nucleus for target gene transcription. Binding of SuFu to GLI is a negative regulation of the pathway as the binding produces GLI-repressor. Presence of ZIC3 will inhibit the production of GLI-repressor and affect the gene transcription process in the nucleus. Repression of the IFT genes will cause disruption of the ciliary structure and function which also will cause laterality defects and HTX. (C) Canonical WNT signaling pathway. WNT binds to its receptor, FZD and co-receptor, LRP5/6. The binding induces dissociation of a complex consisting of APC, AXIN/DVL/GSK3/CK1/β-catenin. The result of the dissociation is the release of β-catenin which is then able to enter the nucleus to regulate target genes transcription by interacting with TCF. ZIC3 binding to TCF has the ability to repress β-catenin, hence affecting the WNT signaling pathway. It is suggested that β-catenin repression will give effect of suppressing the WNT pathway. Another suggested effect is reduction or accumulation of β-catenin. Both lead to failure in forming the dorsal axis, disrupting organ arrangements and cause HTX.
The Hedgehog signaling pathway is another significant component in mammals’ organ development where the Sonic Hedgehog (Shh), Indian-Hedgehog (Ihh) and Desert-Hedgehog (Dhh) are the three main Hedgehog proteins in the pathway.65 The binding of Hedgehog ligands to the PTCH will stop its inhibition on the SMO, hence allowing the downstream pathway. The downstream pathway includes interactions with GLI family (Figure 2(B)), where repressed SMO prevents GLI movement into the nucleus and retains it in the cytoplasm, by binding GLI to SuFu.65,66 The binding of GLI to SuFu does not only prevent target gene transcription by GLI but also leads to production of the GLI protein repressor by proteasome.48,67 ZIC3 interactions with the GLI in the pathway were found to cause alterations to the ratio of activator to repressor of GLI.48 The alterations of the GLI activator and repressor lead to target gene repression such as IFT genes (important for cilia structure and maintenance).67,68 Repression of the IFT genes leads to problems in the cilia structure and function,67,68 hence, causing HTX.
Apart from the Hedgehog signaling pathway, the WNT signaling pathway disruption was also inferred to have caused alterations to the left–right patterning. Asymmetry left–right patterning can be initiated by multiple WNT ligands.48 In the pathway, WNT binds to its receptor and co-receptor (FZD and LRP 5/6, respectively),which then initiates the dissociation of a complex (APC, AXIN/DVL/GSK3/CK1/β-catenin) and releases β-catenin from the complex (Figure 2(C)).69 β-Catenin interacts with TCF to regulate target genes transcription.48 Loss of Wnt3a in the dorsal posterior node, for example, causes aberrant left–right patterning due to delayed expression of certain proteins such as Nodal, Lefty2 and Pitx2 bilaterally in the LPM.70ZIC3 is one of the genes that has been suggested to have an effect on the WNT pathway leading to HTX.48 Fujimi et al. (2012) performed an animal study and discussed WNT signaling pathway suppression by ZIC3 and the effect of ZIC3 interaction in the pathway.71 They hypothesized that WNT signaling suppression by ZIC3 is tissue-specific as the observation of some mammalian cell lines did not show suppression of the WNT signaling.71 It is also hypothesized that ZIC3 interaction with TCF could repress WNT signaling.48 Reduction or overaccumulation of the maternal β-catenin leads to failure in dorsal axis development which is important in determining the arrangement of organs during embryogenesis.72
Dysfunctions in one pathway can influence the others, disrupting the precise coordination needed for proper left–right axis formation.73,74 For instance, variants in genes involved in the Nodal pathway, such as NODAL, ACVR1, and ACVR1B, can lead to abnormal Nodal signaling. This, in turn, affects Hedgehog signaling as Nodal induces the expression of Sonic Hedgehog (SHH) and reciprocally Hedgehog regulates Nodal-related genes.58,73 Concurrently, dysregulation in the canonical WNT pathway can impact the expression of Lefty proteins, key inhibitors of Nodal signaling.58 This disturbs the cross-talk between Nodal and canonical WNT pathways and influences Hedgehog signaling through altered SHH expression.74 The cumulative effects of these dysregulations disrupt the finely tuned interaction among the pathways, leading to the misalignment of internal organs, including the heart, and the manifestation of HTX. This asymmetry is also influenced by cilia activity and their role in creating the left–right organizer flow dynamics.75 It is crucial to note that HTX is a genetically heterogeneous condition, and not all cases can be attributed to variants in these pathways alone, indicating the involvement of other genetic and non-genetic factors in the development of the disorder.
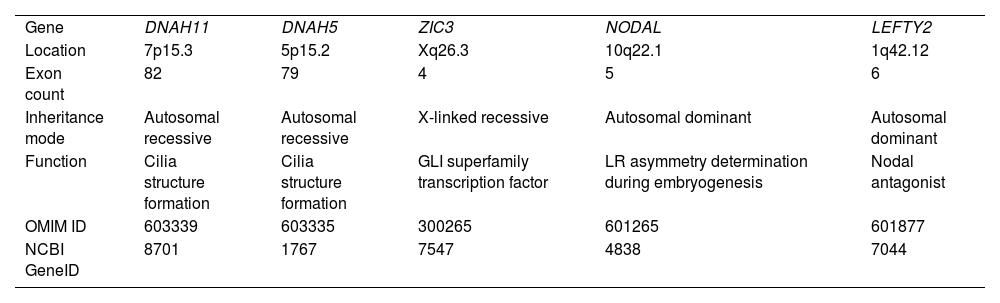
Genes associated with heterotaxy syndromePrevious studies have reported several genetic variants that are associated with HTX.41 HTX may be inherited by different modes depending on the genes that cause the condition.15Table 1 is a summary of the genes discussed in this review that are relevant to HTX.
Summary of the HTX-related genes discussed in this review.
| Gene | DNAH11 | DNAH5 | ZIC3 | NODAL | LEFTY2 |
| Location | 7p15.3 | 5p15.2 | Xq26.3 | 10q22.1 | 1q42.12 |
| Exon count | 82 | 79 | 4 | 5 | 6 |
| Inheritance mode | Autosomal recessive | Autosomal recessive | X-linked recessive | Autosomal dominant | Autosomal dominant |
| Function | Cilia structure formation | Cilia structure formation | GLI superfamily transcription factor | LR asymmetry determination during embryogenesis | Nodal antagonist |
| OMIM ID | 603339 | 603335 | 300265 | 601265 | 601877 |
| NCBI GeneID | 8701 | 1767 | 7547 | 4838 | 7044 |
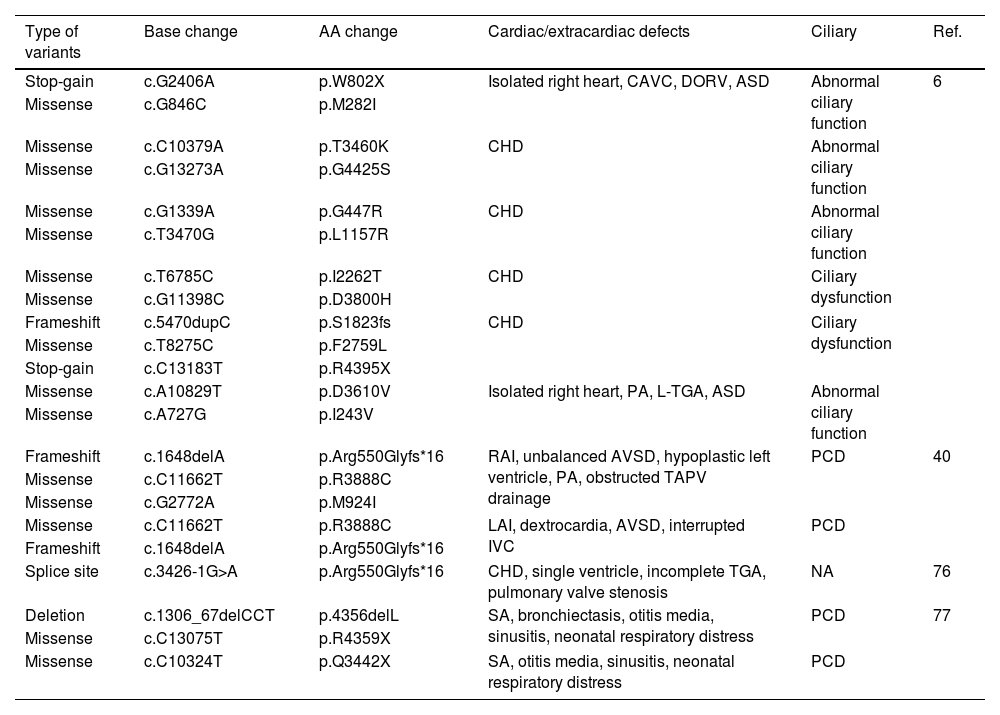
DNAH11 is one of the genes responsible for forming the protein required in cilia structure. Apart from HTX, variants in DNAH11 are also linked to PCD.40 When comparing patients with and without CD, it was discovered that the mutation rate of the gene varied, with the rate of mutation being greater in patients with CD.6DNAH11 variants were predicted to affect splicing and have damaging effects on the protein, potentially leading to the development of heterotaxy and congenital heart defects.41 A study by Liu et al. (2019) found that out of a total of 34 variants, 14 were variants in the DNAH11 gene, which is the highest number of variants found in one gene in the study.6 In a family, two children were found to have the same DNAH11 variants (c.G3426-1A and c.C4306T); however, only the proband was affected with HTX while the other sibling was not.76 According to Xia et al. (2021), this occurrence is attributed by incomplete penetrance or the presence of other variants that inhibit the DNAH11 variants to develop HTX phenotypes.76
The variants are inherited in an autosomal recessive manner where various types of variants such as stop-gain, frameshift, missense, and splice site were reported.6,76Table 2 lists previously reported DNAH11 variants.
DNAH11 variants found to be associated with HTX in previous studies.
| Type of variants | Base change | AA change | Cardiac/extracardiac defects | Ciliary | Ref. |
|---|---|---|---|---|---|
| Stop-gain | c.G2406A | p.W802X | Isolated right heart, CAVC, DORV, ASD | Abnormal ciliary function | 6 |
| Missense | c.G846C | p.M282I | |||
| Missense | c.C10379A | p.T3460K | CHD | Abnormal ciliary function | |
| Missense | c.G13273A | p.G4425S | |||
| Missense | c.G1339A | p.G447R | CHD | Abnormal ciliary function | |
| Missense | c.T3470G | p.L1157R | |||
| Missense | c.T6785C | p.I2262T | CHD | Ciliary dysfunction | |
| Missense | c.G11398C | p.D3800H | |||
| Frameshift | c.5470dupC | p.S1823fs | CHD | Ciliary dysfunction | |
| Missense | c.T8275C | p.F2759L | |||
| Stop-gain | c.C13183T | p.R4395X | |||
| Missense | c.A10829T | p.D3610V | Isolated right heart, PA, L-TGA, ASD | Abnormal ciliary function | |
| Missense | c.A727G | p.I243V | |||
| Frameshift | c.1648delA | p.Arg550Glyfs*16 | RAI, unbalanced AVSD, hypoplastic left ventricle, PA, obstructed TAPV drainage | PCD | 40 |
| Missense | c.C11662T | p.R3888C | |||
| Missense | c.G2772A | p.M924I | |||
| Missense | c.C11662T | p.R3888C | LAI, dextrocardia, AVSD, interrupted IVC | PCD | |
| Frameshift | c.1648delA | p.Arg550Glyfs*16 | |||
| Splice site | c.3426-1G>A | p.Arg550Glyfs*16 | CHD, single ventricle, incomplete TGA, pulmonary valve stenosis | NA | 76 |
| Deletion | c.1306_67delCCT | p.4356delL | SA, bronchiectasis, otitis media, sinusitis, neonatal respiratory distress | PCD | 77 |
| Missense | c.C13075T | p.R4359X | |||
| Missense | c.C10324T | p.Q3442X | SA, otitis media, sinusitis, neonatal respiratory distress | PCD |
ASD: atrial septal defect; CAVC: complete atrioventricular canal; CHD: congenital heart disease; DORV: double outlet right ventricle; IVC: inferior vena cava; LAI: left atrial isomerism; L-TGA: Levo-transposition of the great arteries; NA: not announced; PA: pulmonary atresia; PCD: primary ciliary dyskinesia; RAI: right atrial isomerism; SA: situs ambiguous; TAPV: total anomalous pulmonary venous; TGA: transposition of the great arteries.
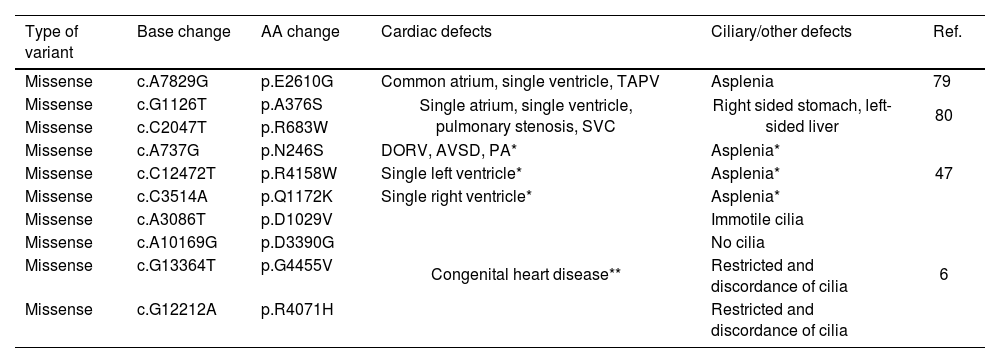
Dynein axonemal heavy chain 5 is another gene that is associated with HTX due to its function in encoding proteins for the ciliary structure, specifically the outer dynein arm.78 It is mainly associated with PCD, and PCD caused by DNAH5 has been associated with laterality defects including situs ambiguous or HTX.78 The gene is inherited in an autosomal recessive manner and trans-heterozygosity of DNAH5 with DNAH6 and DNAH11 has been observed to cause HTX (Table 3).6,47
DNAH5 variants found to be associated with HTX in previous studies.
| Type of variant | Base change | AA change | Cardiac defects | Ciliary/other defects | Ref. |
|---|---|---|---|---|---|
| Missense | c.A7829G | p.E2610G | Common atrium, single ventricle, TAPV | Asplenia | 79 |
| Missense | c.G1126T | p.A376S | Single atrium, single ventricle, pulmonary stenosis, SVC | Right sided stomach, left-sided liver | 80 |
| Missense | c.C2047T | p.R683W | |||
| Missense | c.A737G | p.N246S | DORV, AVSD, PA* | Asplenia* | 47 |
| Missense | c.C12472T | p.R4158W | Single left ventricle* | Asplenia* | |
| Missense | c.C3514A | p.Q1172K | Single right ventricle* | Asplenia* | |
| Missense | c.A3086T | p.D1029V | Congenital heart disease** | Immotile cilia | 6 |
| Missense | c.A10169G | p.D3390G | No cilia | ||
| Missense | c.G13364T | p.G4455V | Restricted and discordance of cilia | ||
| Missense | c.G12212A | p.R4071H | Restricted and discordance of cilia |
AVSD: atrioventricular septal defects; DORV: double outlet right ventricle; PA: pulmonary atresia; SVC: superior vena cava; TAPV: total anomalous pulmonary venous drainage.
Zinc finger in cerebellum 3 is a gene from the GLI superfamily that acts as a transcription factor during neurogenesis, and previous studies using animal models suggested that reduced expression and/or activity of ZIC3 protein during early embryogenesis affects the laterality patterning.81ZIC3 is a gene with significant evidence of its pathogenicity in causing HTX, where at least 36 ZIC3 variants have been discovered to be pathogenic since the first ZIC3 variant was found in 1997.82 Variants in the ZIC3 gene have been shown to have a significant impact on heterotaxy, particularly in male patients with abnormal arrangement of thoracic and visceral organs.81 These variants, which are predominantly located in the zinc finger domains or result in truncation before these domains, have been found to cause abnormal trafficking of mutated ZIC3 proteins.83 Functional analysis in zebrafish models has further confirmed the pathogenic nature of these variants, with frameshift variants showing full non-functionality and a missense variant showing partial activity compared to the wild-type.84 Additionally, variants in the N-terminal and zinc finger-binding domains of ZIC3 have been found to result in the loss of reporter gene transactivation and aberrant cytoplasmic localization, respectively.82
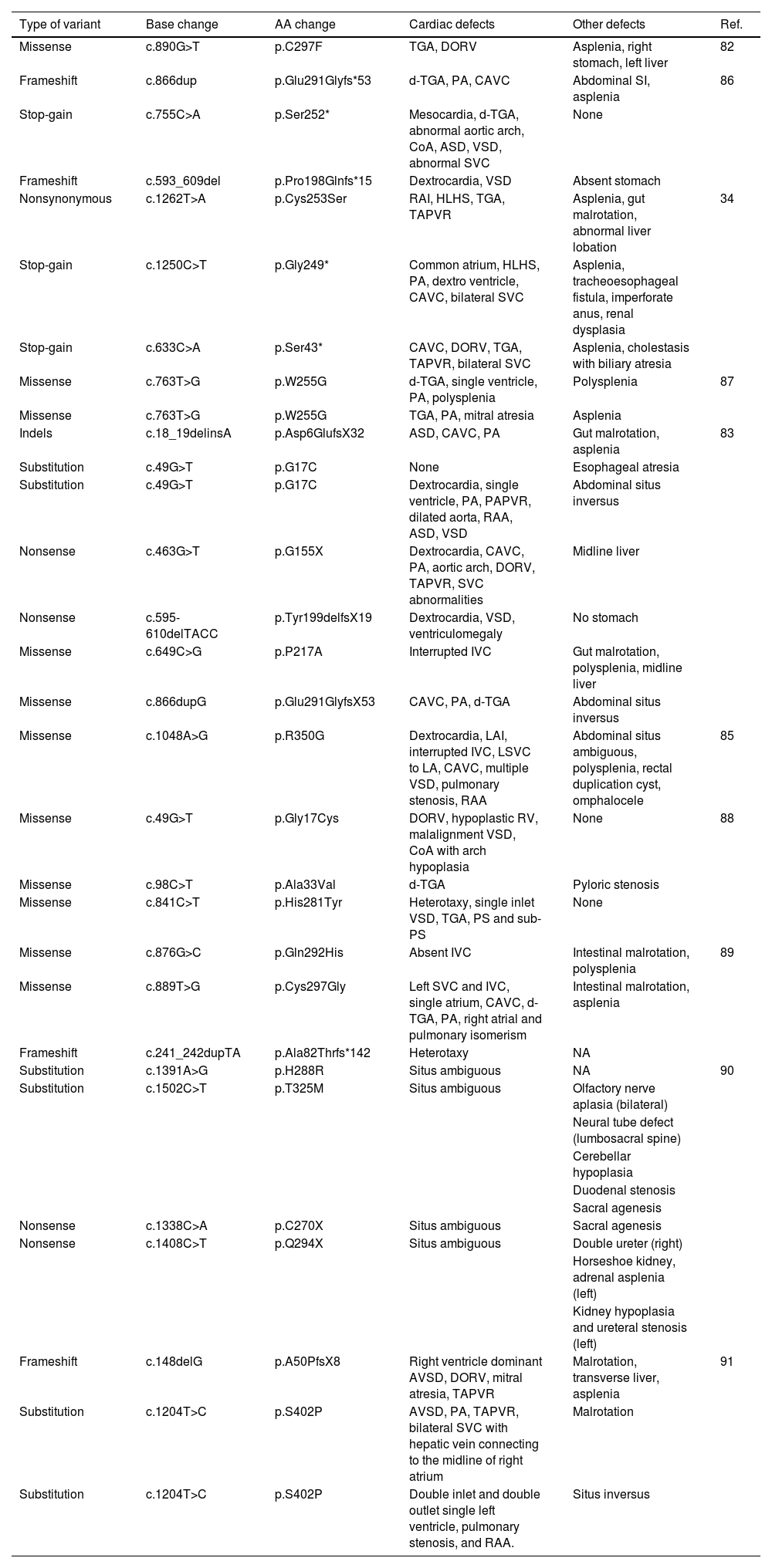
A functional investigation of a novel hemizygous mutation (c.890G>T, p.C297F) in the ZIC3 gene revealed that this specific mutation reduces the transcriptional activity of ZIC3. It affects the binding affinity of the GLI-binding site and leads to abnormal cellular localization when introduced into transfected cells. In addition, the variant (p.C297F) was found to affect protein structure and DNA-binding ability.82 Other than that, ZIC3 is the earliest gene associated with HTX reported and the only gene to be inherited by X-linked inheritance of HTX.83 D’alessandro et al. (2011) reported that the women in the studied family were carriers with no phenotypic manifestations, while several male family members manifested congenital heart diseases.85 One of the male family members was also found to carry the variant; however, no morphological defects of the organs were observed, which was confirmed to be due to incomplete penetrance of the ZIC3 gene variant.85 Five out of 100 familial HTX was found to be caused by ZIC3 variants, while 1 out of 100 were sporadic cases.35,85 Previously reported ZIC3 variants are listed in Table 4.
ZIC3 variants found to be associated with HTX in previous studies.
| Type of variant | Base change | AA change | Cardiac defects | Other defects | Ref. |
|---|---|---|---|---|---|
| Missense | c.890G>T | p.C297F | TGA, DORV | Asplenia, right stomach, left liver | 82 |
| Frameshift | c.866dup | p.Glu291Glyfs*53 | d-TGA, PA, CAVC | Abdominal SI, asplenia | 86 |
| Stop-gain | c.755C>A | p.Ser252* | Mesocardia, d-TGA, abnormal aortic arch, CoA, ASD, VSD, abnormal SVC | None | |
| Frameshift | c.593_609del | p.Pro198Glnfs*15 | Dextrocardia, VSD | Absent stomach | |
| Nonsynonymous | c.1262T>A | p.Cys253Ser | RAI, HLHS, TGA, TAPVR | Asplenia, gut malrotation, abnormal liver lobation | 34 |
| Stop-gain | c.1250C>T | p.Gly249* | Common atrium, HLHS, PA, dextro ventricle, CAVC, bilateral SVC | Asplenia, tracheoesophageal fistula, imperforate anus, renal dysplasia | |
| Stop-gain | c.633C>A | p.Ser43* | CAVC, DORV, TGA, TAPVR, bilateral SVC | Asplenia, cholestasis with biliary atresia | |
| Missense | c.763T>G | p.W255G | d-TGA, single ventricle, PA, polysplenia | Polysplenia | 87 |
| Missense | c.763T>G | p.W255G | TGA, PA, mitral atresia | Asplenia | |
| Indels | c.18_19delinsA | p.Asp6GlufsX32 | ASD, CAVC, PA | Gut malrotation, asplenia | 83 |
| Substitution | c.49G>T | p.G17C | None | Esophageal atresia | |
| Substitution | c.49G>T | p.G17C | Dextrocardia, single ventricle, PA, PAPVR, dilated aorta, RAA, ASD, VSD | Abdominal situs inversus | |
| Nonsense | c.463G>T | p.G155X | Dextrocardia, CAVC, PA, aortic arch, DORV, TAPVR, SVC abnormalities | Midline liver | |
| Nonsense | c.595-610delTACC | p.Tyr199delfsX19 | Dextrocardia, VSD, ventriculomegaly | No stomach | |
| Missense | c.649C>G | p.P217A | Interrupted IVC | Gut malrotation, polysplenia, midline liver | |
| Missense | c.866dupG | p.Glu291GlyfsX53 | CAVC, PA, d-TGA | Abdominal situs inversus | |
| Missense | c.1048A>G | p.R350G | Dextrocardia, LAI, interrupted IVC, LSVC to LA, CAVC, multiple VSD, pulmonary stenosis, RAA | Abdominal situs ambiguous, polysplenia, rectal duplication cyst, omphalocele | 85 |
| Missense | c.49G>T | p.Gly17Cys | DORV, hypoplastic RV, malalignment VSD, CoA with arch hypoplasia | None | 88 |
| Missense | c.98C>T | p.Ala33Val | d-TGA | Pyloric stenosis | |
| Missense | c.841C>T | p.His281Tyr | Heterotaxy, single inlet VSD, TGA, PS and sub-PS | None | |
| Missense | c.876G>C | p.Gln292His | Absent IVC | Intestinal malrotation, polysplenia | 89 |
| Missense | c.889T>G | p.Cys297Gly | Left SVC and IVC, single atrium, CAVC, d-TGA, PA, right atrial and pulmonary isomerism | Intestinal malrotation, asplenia | |
| Frameshift | c.241_242dupTA | p.Ala82Thrfs*142 | Heterotaxy | NA | |
| Substitution | c.1391A>G | p.H288R | Situs ambiguous | NA | 90 |
| Substitution | c.1502C>T | p.T325M | Situs ambiguous | Olfactory nerve aplasia (bilateral) | |
| Neural tube defect (lumbosacral spine) | |||||
| Cerebellar hypoplasia | |||||
| Duodenal stenosis | |||||
| Sacral agenesis | |||||
| Nonsense | c.1338C>A | p.C270X | Situs ambiguous | Sacral agenesis | |
| Nonsense | c.1408C>T | p.Q294X | Situs ambiguous | Double ureter (right) | |
| Horseshoe kidney, adrenal asplenia (left) | |||||
| Kidney hypoplasia and ureteral stenosis (left) | |||||
| Frameshift | c.148delG | p.A50PfsX8 | Right ventricle dominant AVSD, DORV, mitral atresia, TAPVR | Malrotation, transverse liver, asplenia | 91 |
| Substitution | c.1204T>C | p.S402P | AVSD, PA, TAPVR, bilateral SVC with hepatic vein connecting to the midline of right atrium | Malrotation | |
| Substitution | c.1204T>C | p.S402P | Double inlet and double outlet single left ventricle, pulmonary stenosis, and RAA. | Situs inversus |
ASD: atrial septal defect; CAVC: complete atrioventricular canal; CoA: coarctation of the aorta; DORV: double outlet right ventricle; d-TGA: dextro-transposition of the great arteries; HLHS: hypoplastic left heart syndrome; IVC: inferior vena cava; LAI: left atrial isomerism; LSVC: left superior vena cava; NA: not announced; PA: pulmonary atresia; PAPVR: partial anomalous pulmonary venous return; PS: pulmonary stenosis; RAA: right aortic arch; RAI: right atrial isomerism; RV: right ventricle; SVC: superior vena cava; TAPVR: total anomalous pulmonary venous return; TGA: transposition of the great arteries; VSD: ventricle septal defect.
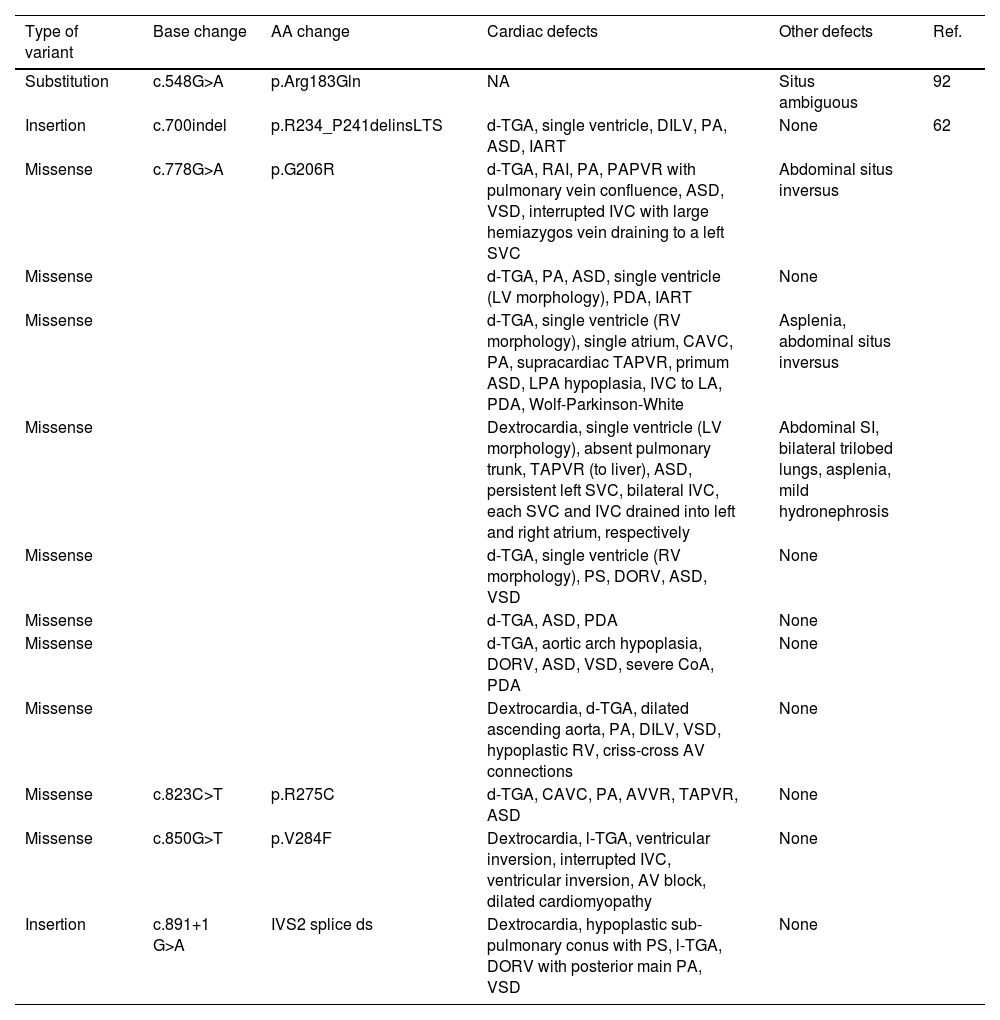
NODAL protein is a secreted molecule from the Nodal signaling pathway. It is a regulator that belongs to the TGF-β family, hence it plays a significant role in the determination of left–right asymmetry in vertebrates.62 The expression of NODAL is primarily symmetry but progresses to asymmetrical toward the left side of the LPM, and the gene expression is highly conserved.92 Multiple studies have identified missense variants, in-frame insertion/deletion, and splice site variants in the NODAL gene in patients with heterotaxy and/or isolated cardiovascular malformations (CVM).62 Functional analyses have shown that these NODAL variants exhibit impaired signaling, leading to decreased activation of artificial reporters and reduced induction of SMAD2 phosphorylation and nuclear import.93 Additionally, a study on mouse mutants with a mutation in the MEGF8 gene, which is involved in left–right patterning, showed that Nodal signaling failed to be propagated to the left LPM, resulting in heterotaxy and CHD.94 These findings suggest that mutations and rare deleterious variants in NODAL can contribute to the development of sporadic human heterotaxy.95 A study by Mohapatra et al. (2009) found seven variants where four are missense, while one is in-frame insertion/deletion and two are splice site variants.62 Previously reported NODAL variants are listed in Table 5.
NODAL variants found to be associated with HTX in previous studies.
| Type of variant | Base change | AA change | Cardiac defects | Other defects | Ref. |
|---|---|---|---|---|---|
| Substitution | c.548G>A | p.Arg183Gln | NA | Situs ambiguous | 92 |
| Insertion | c.700indel | p.R234_P241delinsLTS | d-TGA, single ventricle, DILV, PA, ASD, IART | None | 62 |
| Missense | c.778G>A | p.G206R | d-TGA, RAI, PA, PAPVR with pulmonary vein confluence, ASD, VSD, interrupted IVC with large hemiazygos vein draining to a left SVC | Abdominal situs inversus | |
| Missense | d-TGA, PA, ASD, single ventricle (LV morphology), PDA, IART | None | |||
| Missense | d-TGA, single ventricle (RV morphology), single atrium, CAVC, PA, supracardiac TAPVR, primum ASD, LPA hypoplasia, IVC to LA, PDA, Wolf-Parkinson-White | Asplenia, abdominal situs inversus | |||
| Missense | Dextrocardia, single ventricle (LV morphology), absent pulmonary trunk, TAPVR (to liver), ASD, persistent left SVC, bilateral IVC, each SVC and IVC drained into left and right atrium, respectively | Abdominal SI, bilateral trilobed lungs, asplenia, mild hydronephrosis | |||
| Missense | d-TGA, single ventricle (RV morphology), PS, DORV, ASD, VSD | None | |||
| Missense | d-TGA, ASD, PDA | None | |||
| Missense | d-TGA, aortic arch hypoplasia, DORV, ASD, VSD, severe CoA, PDA | None | |||
| Missense | Dextrocardia, d-TGA, dilated ascending aorta, PA, DILV, VSD, hypoplastic RV, criss-cross AV connections | None | |||
| Missense | c.823C>T | p.R275C | d-TGA, CAVC, PA, AVVR, TAPVR, ASD | None | |
| Missense | c.850G>T | p.V284F | Dextrocardia, l-TGA, ventricular inversion, interrupted IVC, ventricular inversion, AV block, dilated cardiomyopathy | None | |
| Insertion | c.891+1 G>A | IVS2 splice ds | Dextrocardia, hypoplastic sub-pulmonary conus with PS, l-TGA, DORV with posterior main PA, VSD | None |
ASD: atrial septal defect; AV: atrioventricular; AVVR: atrioventricular valve regurgitation; CAVC: complete atrioventricular canal; CoA: coarctation of the aorta; DILV: double inlet left ventricle; DORV: double outlet right ventricle; d-TGA: dextro-transposition of the great artery; IART: intraarterial re-entrant tachycardia; IVC: inferior vena cava; LPA: left pulmonary artery; LV: left ventricle; l-TGA: Levo-transposition of the great arteries; NA: not announced; PA: pulmonary atresia; PAPVR: partial anomalous pulmonary venous return; PDA: patent ductus arteriosus; PS: pulmonary stenosis; RAI: right atrial isomerism; RV: right ventricle; SVC: superior vena cava; TAPVR: total anomalous pulmonary venous return; VSD: ventricle septal defect.
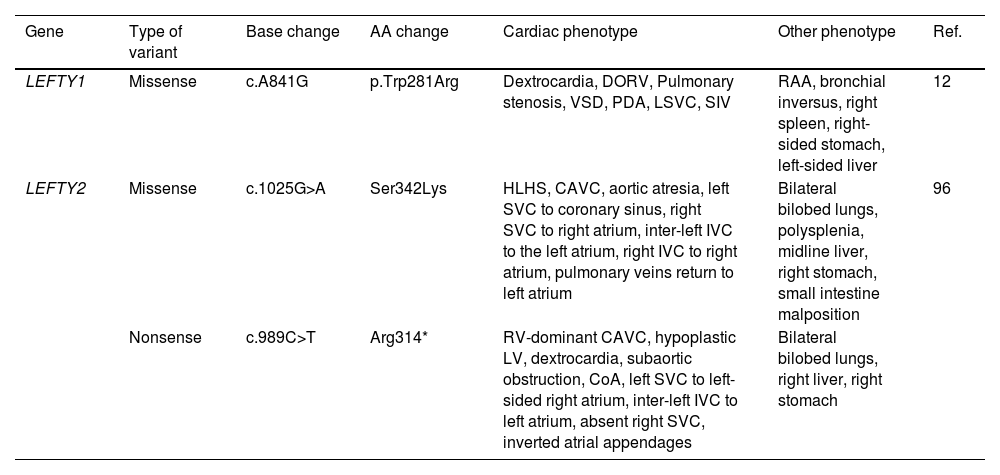
There are two LEFTY genes (LEFTY1 and LEFTY2), which are involved in Nodal signaling from node to the LPM.91LEFTY1 and LEFTY2 genes also belong to the TGF-β superfamily.93 They are NODAL antagonists in the Nodal signaling pathway, where they negatively regulate NODAL expression by blocking receptor complex activation.60 LEFTY1 is expressed in the midline of LPM while LEFTY2 is expressed within the left LPM, where both retain the NODAL signaling suppression.78 In one study, rare copy number variants (CNVs) were identified in patients with heterotaxy, and functional testing in zebrafish models revealed that downregulation of LEFTY disrupted cardiac looping and abnormal expression of left–right patterning markers.62 A nonsynonymous heterozygous variant pair of LEFTY1 (c.A841G) and TTC40 candidate gene were discovered in a CNV study.12 However, most LEFTY1 gene studies focus on gene expressions. Animal studies have revealed that the absence of Lefty1 affects both Nodal and Lefty2 genes expressions in which they are expressed on both sides of the lateral plates instead of only on the left side.64,91 Apart from LEFTY1, a study by Kosaki et al. (1999) found two sporadic variants (c.1025G>A and c.989C>T) of LEFTY2 cases that phenotypically fit the heterotaxy description.96 The variants were discovered to be in the cysteine knot region, which is critical as it directly implicates the binding of receptors.96 Two other LEFTY2 variants (c.-225C>T, ClinVarID: 295978, and c.-222C>A, ClinVarID: 295977) associated with HTX have been reported in the ClinVar database by Illumina, nevertheless no publications have reported these variants.97,98LEFTY2 is transcriptionally regulated by a complex which includes p300, SMAD2, and CITED2. Su et al. (2013), suggested that downregulation of LEFTY2 might play a role in the onset of VSD partly by triggering abnormal activation of the NODAL-PITX2C pathway.99 Additionally, the data reveal that the histone acetyltransferase p300 contributes to the activation of LEFTY2 by promoting hyperacetylation of histone H4 at the promoter region of LEFTY2. Previously reported LEFTY1 and LEFTY2 variants are listed in Table 6.
LEFTY1 and LEFTY2 variants found to be associated with HTX in previous studies.
| Gene | Type of variant | Base change | AA change | Cardiac phenotype | Other phenotype | Ref. |
|---|---|---|---|---|---|---|
| LEFTY1 | Missense | c.A841G | p.Trp281Arg | Dextrocardia, DORV, Pulmonary stenosis, VSD, PDA, LSVC, SIV | RAA, bronchial inversus, right spleen, right-sided stomach, left-sided liver | 12 |
| LEFTY2 | Missense | c.1025G>A | Ser342Lys | HLHS, CAVC, aortic atresia, left SVC to coronary sinus, right SVC to right atrium, inter-left IVC to the left atrium, right IVC to right atrium, pulmonary veins return to left atrium | Bilateral bilobed lungs, polysplenia, midline liver, right stomach, small intestine malposition | 96 |
| Nonsense | c.989C>T | Arg314* | RV-dominant CAVC, hypoplastic LV, dextrocardia, subaortic obstruction, CoA, left SVC to left-sided right atrium, inter-left IVC to left atrium, absent right SVC, inverted atrial appendages | Bilateral bilobed lungs, right liver, right stomach |
CAVC: complete atrioventricular canal: CoA: coarctation of the aorta: DORV: double outlet right ventricle: HLHS: hypoplastic left heart syndrome: IVC: inferior vena cava: LSVC: left superior vena cava: LV: left ventricle: PDA: patent ductus arteriosus: RAA: right aortic arch: RV: right ventricle: SIV: superior-inferior ventricle: SVC: superior vena cava: VSD: ventricle septal defect.
Based on NCBI NIH Genetic Testing Registry, HTX gene panel for diagnosis includes 10 genes, including NKX2-5, GDF1, GJA1, NODAL, CFC1, LEFTY2, ZIC3, CRELD1, FOXH1 and ACVR2B.100DNAI1, MMP21, PKD1L1, and HYDIN, are other known PCD genes associated with HTX besides those discussed above, while ROCK2, ISL1 and SMAD2 are newly found candidate genes.85
According to Litonjua and Celedón (2006), the definition of candidate gene is a gene that is suggested to have a link to a particular illness or trait by the discovery of either the protein products or chromosomal location.101 Further investigations are required to understand these genes better.
A study conducted using CNVs analysis suggested two candidate genes which are dynein axonemal heavy chain 10 (DNAH10) and ring finger protein 115 (RNF115), where DNAH10 was proposed to disrupt ciliary function as it affects notochord, while RNF115 affects the laterality signaling pathways.12 Six novel candidate genes were found in a study of 25 HTX patients, i.e., FLNA, ITGA1, PCNT, KIF7, GLI1, and KMT2D.102KIF7 and FLNA are indirectly involved in the ciliary movement as KIF7 encodes for cilia associated protein in the Shh signaling pathway, while FLNA encodes for actin-binding proteins involved in cytoskeleton function.102 Apart from the genes discussed in the review, homozygous missense mutation of SHROOM3 gene (c.G179T) was reported in a study; however, the variant cannot be found in the databases.103 In addition, PFKP is among the top HTX candidate genes with proof of its effect in an animal model, Xenopus laevis.13PITX2 expression is important for the formation of the heart, and its abnormal expression has been linked to CHDs.104 Studies in animal models further support the connection of abnormal expression of PITX2 with HTX.105,106
Current limitations and future research directionsThe quest to unravel the genetic underpinnings of HTX and CHD is a challenging yet rewarding endeavor. At present, our understanding is substantial but constrained by several limitations. Predominantly, genetic studies exhibit an inclination toward populations of European ancestry, thereby narrowing the diversity and generalizability of the findings. Most identified genetic factors are inferred from association studies, which primarily establish a correlation rather than causation. Establishing causality requires extensive functional studies, which are often impeded by time and resource constraints. The genetics of HTX and CHD represent a complex interplay of multiple genes and environmental factors, adding layers of intricacy to the understanding of these conditions. Additionally, the role of epigenetic factors in HTX and CHD is relatively nascent, necessitating further exploration.
Despite these hurdles, the prospective avenues in HTX and CHD genetic research hold immense promise. Emerging methodologies, including genome-wide association studies and whole exome sequencing, are expected to uncover novel genetic factors. An in-depth exploration of gene–gene and gene–environment interactions is expected to provide a more comprehensive understanding of these conditions. One area of particular interest is the genetic basis of CHD in the context of HTX, which may elucidate why certain individuals develop CHD while others do not. Progress in our understanding could pave the way for targeted therapies and precision medicine, revolutionizing the treatment landscape. The role of epigenetic factors, such as DNA methylation, histone modifications and noncoding RNA activity, represents another promising research frontier. Functional studies of genetic variants associated with HTX and CHD are expected to shed light on the mechanisms through which these variants alter cellular functions and contribute to disease pathogenesis. The path forward, while fraught with challenges, is replete with opportunities for significant breakthroughs and advancements.
ConclusionHeterotaxy syndrome is commonly associated with complex CHD that accounts for significant morbidity and mortality. Currently, multiple HTX-related genes are being discovered and their association elucidated, including their inheritance patterns of either autosomal recessive, autosomal dominant, or X-linked inheritance. HTX's etiology is hard to understand due to the multiple genes that can possibly be associated with HTX. The phenotypic defects between the RAI and LAI may be determined generally; however, further research should be conducted as there are overlapping cardiac defects among them. Disruption of ciliary function has been observed to have caused the abnormal expression of certain proteins during embryogenesis, leading to abnormal organ arrangement. Apart from that, signaling pathway disruptions due to gene variants are inferred to have been one of the HTX etiology. Nevertheless, further studies are required to validate them and consider the possibility of environmental factors being involved.
Conflict of interestNo potential conflict of interest was reported by the authors.
We would like to thank Ms. Ana Leticia Gomes for her invaluable assistance in translating the English language abstract into Portuguese.
