





We report the case of a 52-year-old man who presented to our emergency department (ED) after three episodes of syncope in the seven hours before admission. During his stay in the ED he had recurrent ventricular tachycardia (VT) requiring external electrical cardioversion. A 12-lead electrocardiogram (ECG) showed a short QT (SQT) interval (270 ms, QTc 327 ms), with frequent R-on-T extrasystoles triggering sustained polymorphic VT. After exclusion of other precipitating causes, the patient was diagnosed as having SQT syndrome (SQTS) according to the Gollob criteria. To our knowledge, this is the first known documentation of an SQT-caused arrhythmic episode on a 12-lead ECG, as well as the first reported case of SQTS in Portugal. The patient received an implantable cardioverter-defibrillator and was discharged. At a follow-up assessment 14 months later he was symptom-free, interrogation of the device showed no arrhythmic events, and the ECG showed a QT interval of 320 ms (QTc 347 ms).
Os autores apresentam o caso de um homem com 52 anos que recorreu ao Serviço de Urgência (SU) por três episódios sincopais ocorridos nas últimas sete horas pré-admissão hospitalar. Durante a estadia no SU, objetivaram-se períodos de taquicardia ventricular (TV) recorrente, com necessidade de cardioversão elétrica externa, tendo o doente sido transferido para a unidade de cuidados intensivos cardíacos para monitorização e orientação clínica. O electrocardiograma de 12 derivações (ECG) registado no contexto de um dos episódios revelava um intervalo QT curto (260 ms, QTc 327 ms) em ritmo sinusal, com fenómeno de R-on-T desencadeando TV polimórfica mantida. Após exclusão de causas precipitantes, o doente foi classificado como tendo Síndrome QT Curto (SQTC), de acordo com os critérios de Gollob. Tanto quanto é do nosso conhecimento, esta é a primeira documentação no ECG de um episódio arrítmico causado por SQTC e a primeira descrição de um caso de SQTC em Portugal. Foi submetido a implantação de cardioversor-desfibrilhador implantável (CDI) monocameral e teve alta hospitalar. No seguimento aos 14 meses pós-implantação, o doente negava recorrência da sintomatologia, a interrogação do CDI não mostrou novos episódios arrítmicos, e no ECG persistia um intervalo QT de 320 ms (QTc 347 ms).
electrocardiogram
emergency department
implantable cardioverter-defibrillator
premature ventricular contractions
polymorphic ventricular tachycardia
corrected QT
short QT
short QT syndrome
ventricular tachycardia
A 52-year-old man presented to our emergency department (ED) due to three episodes of sudden transient loss of consciousness in the seven hours before hospital admission. The first event occurred at 2:00 am, while the patient was lying in bed, recurring at 4:00 am and 7:00 am. He reported malaise, but denied prodromes, chest pain, palpitations or dyspnea. His wife, who witnessed the episodes, described them as lasting for about five minutes, with spontaneous and complete recovery. There was no evidence of seizure-like activity.
The patient had no history of known cardiovascular disease and no recollection of previous episodes of syncope. His father died at 38 years of age due to sudden death of undetermined origin, with no other relevant family history. His medical history was notable for depression (managed at an outpatient clinic), duodenal peptic ulcer (five years previously), and hypercholesterolemia. His regular medication consisted of mirtazapine 30 mg daily, alprazolam 0.5 mg daily, bupropion 300 mg daily and fenofibrate 200 mg daily. He had no clinical allergies and denied smoking, alcohol intake or any kind of addiction.
Physical examination showed a well-nourished man with no external signs of distress. At admission, he was afebrile, with blood pressure of 112/65 mmHg, heart rate of 76 bpm and oxygen saturation of 98% by pulse oximetry. Cardiac examination showed an irregular heart rhythm with no murmurs. The rest of the physical examination was unremarkable.
During his assessment in the ED, he suffered a new episode of syncope with spontaneous recovery, and cardiac monitoring during the event showed a polymorphic ventricular tachycardia (PVT). Despite intravenous infusion of amiodarone and magnesium sulphate, there was recurrence of the cardiac arrhythmia requiring a total of five external electrical cardioversions. A 12-lead electrocardiogram (ECG) recorded during one such event is presented in Figure 1.
A blood chemistry panel showed normal serum potassium and magnesium, with no hypocalcemia. Cardiac biomarkers were negative, and other laboratory investigations were unremarkable. The patient was transferred to our cardiac intensive care unit for further assessment and treatment.
Careful review of ECG records (Figures 1 and 2) showed a short QT interval (280 ms, QTc 329 ms according to the Bazett formula). Given the setting of a PVT storm coronary angiography was performed, which ruled out coronary artery disease or anomalous coronary origin (Figures 3 and 4), with normal wall motion and left ventricular ejection fraction (70%). Transthoracic echocardiographic examination showed a normal heart with good left ventricular systolic function and no signs of structural disease.
 ECG recorded during a syncopal episode shows sinus rhythm with a heart rate of 83 bpm. The tracing shows a shortened QT interval (320 ms) with frequent premature ventricular contractions (PVCs) causing R-on-T extrasystoles (red arrow). One
ECG recorded during a syncopal episode shows sinus rhythm with a heart rate of 83 bpm. The tracing shows a shortened QT interval (320 ms) with frequent premature ventricular contractions (PVCs) causing R-on-T extrasystoles (red arrow). One This 12-lead ECG recorded during a syncopal episode shows sinus rhythm with a heart rate of 83 bpm. The tracing shows a shortened QT interval (320 ms) with frequent premature ventricular contractions (PVCs) causing R-on-T extrasystoles (red arrow). One PVC (black arrow) triggers polymorphic ventricular tachycardia which caused loss of consciousness.
 QTc interval according to the Bazett formula12 is 327 ms.' title='Electrocardiographic recording, lead V2, showing shortened QT interval (270 ms) with RR interval of 0.68 s. The
QTc interval according to the Bazett formula12 is 327 ms.' title='Electrocardiographic recording, lead V2, showing shortened QT interval (270 ms) with RR interval of 0.68 s. The Electrocardiographic recording, lead V2, showing shortened QT interval (270 ms) with RR interval of 0.68 s. The QTc interval according to the Bazett formula12 is 327 ms.

 ECG recording at follow-up.' title='
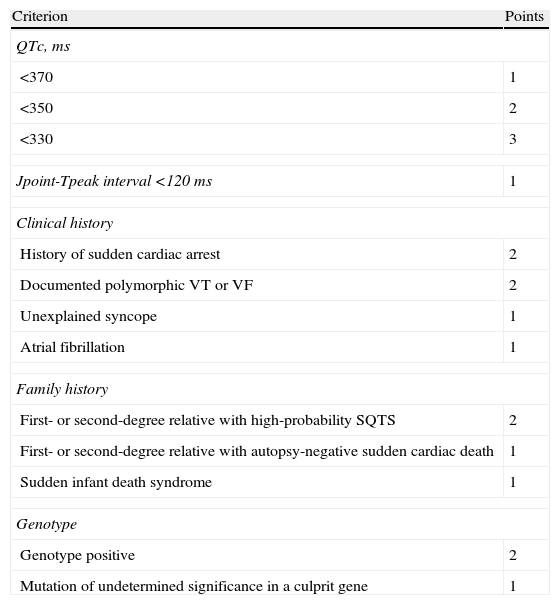
ECG recording at follow-up.' title='In the absence of other causes, a presumptive diagnosis of short QT syndrome (SQTS) was made, and quinidine tablets were requested from the hospital pharmacy. The patient was classified as high-probability SQTS according to the Gollob criteria,1 with a score of 5 (Table 1).
Proposed diagnostic criteria for short QT syndrome.
| Criterion | Points |
| QTc, ms | |
| <370 | 1 |
| <350 | 2 |
| <330 | 3 |
| Jpoint-Tpeak interval <120 ms | 1 |
| Clinical history | |
| History of sudden cardiac arrest | 2 |
| Documented polymorphic VT or VF | 2 |
| Unexplained syncope | 1 |
| Atrial fibrillation | 1 |
| Family history | |
| First- or second-degree relative with high-probability SQTS | 2 |
| First- or second-degree relative with autopsy-negative sudden cardiac death | 1 |
| Sudden infant death syndrome | 1 |
| Genotype | |
| Genotype positive | 2 |
| Mutation of undetermined significance in a culprit gene | 1 |
QTc: corrected QT; SQTS: short QT syndrome; VF: ventricular fibrillation; VT: ventricular tachycardia. High-probability SQTS: 4 points; intermediate-probability SQTS: 3 points; low-probability SQTS: 2 points.
Adapted from Gollob et al.1
In-patient 24-hour Holter monitoring was performed and showed infrequent dimorphic premature ventricular contractions (PVCs), with no repetitive patterns. An electrophysiologic study was performed and was notable for the induction of self-limited PVT with hemodynamic collapse (induction protocol: S1 400/S2 200 ms, pacing at the right ventricular apex). The measured right ventricular refractory period was shortened (200 ms). No attempt was made to induce atrial fibrillation. The measured atrial refractory period was 350 ms in the right atrium and 200 ms in the left atrium (as measured in the distal coronary sinus).
In accordance with the ACCF/AHA/HRS guidelines for device-based therapy,2 the patient received an implantable cardioverter-defibrillator (ICD) as secondary prevention indicated for SQTS, repeated syncopes and inducible PVT. There were no procedural complications and he was discharged on his previous medication and quinidine.
At a follow-up visit 14 months later, the patient was feeling well and had had no new episodes of syncope or palpitations. Physical examination was unremarkable. ICD device interrogation showed no shocks or arrhythmic events, and repeat ECG assessment (Figure 4) showed a QT interval of 320 ms (QTc 347 ms).
Genetic testing was performed, and screening of the KCNJ2, KCNH2 and KCNQ1 genes by polymerase chain reaction was negative for short deletions or missense mutations.
To the authors’ knowledge, this is the first reported case of STQS in Portugal, which is in agreement with the very low prevalence of this condition. As an index case, ECG screening of family members is underway.
DiscussionSudden cardiac death is one of the leading causes of death in the industrialized world, and ventricular arrhythmias have long been recognized as one of the causative mechanisms. While these usually occur in the setting of structural heart disease, they may also be a consequence of an acute process or an inherited predisposition of an individual's cardiac tissue to life-threatening arrhythmias.
These primary cardiac electrical diseases, although uncommon, are usually detected in younger3 and otherwise healthy individuals, often with tragic consequences.
SQTS is a recently described entity included in the group of inherited primary electric disorders. As originally proposed by Gussak in 2000,4 it involved the association of a significantly shortened QTc interval on the surface ECG with atrial fibrillation and sudden cardiac death.
Since its description in two families,5 our understanding of this rare entity has evolved, but there are still many questions to answer, particularly regarding its incidence and prognosis.
Our patient was initially assessed in the ED with a clinical diagnosis of syncope. While this is a common complaint in the ED, representing up to 3% of all visits, its causes range from benign vasovagal reactions to life-threatening cardiac arrhythmias. In this clinical setting, the presentation of recurrent syncope at rest, without known precipitating events, suggested a potentially serious underlying process.
After documentation of an arrhythmic cause, the patient's severe electrical instability could have been associated with an acute myocardial ischemic process, but this was ruled out by persistently negative serum troponin and a normal coronary angiogram. In the absence of other precipitating causes, with ECGs showing a shortened QT interval without structural heart disease, a presumptive diagnosis of SQTS was made. To our knowledge, this is the first known documentation on a 12-lead ECG of R-on-T extrasystoles in a short QT-related arrhythmic episode.
In contrast with long QT interval, which may be brought on by external factors, including certain drugs, there is no known association between medication and a shortened QT interval. However, due to an overlap of QTc values between healthy population cohorts and patients with SQTS,6,7 hyperkalemia, hypo- or hypercalcemia, sinus tachycardia, hypothermia, acidosis or digoxin therapy must be excluded. A case of short QT related to prolonged resuscitation efforts and hypothermia has also been described,8 but this was not the case in our patient.
Similarly to the long QT syndrome, there appears to be significant overlap of QTc values between healthy population cohorts and patients with SQTS.6,7 This suggests that an abbreviated QT interval is not in itself the cause of an increased risk of arrhythmia, but may be a surface surrogate for the arrhythmogenic substrate in SQTS patients. As such, a diagnosis of short QT syndrome should only be considered following careful assessment of the 12-lead ECG, in an appropriate clinical setting (history of syncope or aborted sudden cardiac death, positive family history), as suggested by the Gollob score.
As the QT interval is heart-rate dependent, use of the Bazett formula allows estimation of the real QT interval (as measured at 60 bpm) at any heart rate. Some caution must be used in its application, as the formula has been reported to undercorrect (yielding erroneously high QTc intervals) at heart rates above 90 bpm.14 However, measuring the QT interval at a heart rate of 60 bpm is not always feasible in clinical practice as it may require the use of beta-blockers or Holter monitoring, and the Bazett formula remains an acceptably simple and effective tool at the bedside.
The described cellular mechanism behind idiopathic short QT interval is usually related to gain-of-function mutations in potassium channels; these lead to a shortened action potential duration through accelerated cardiomyocyte repolarization. So far, six different genotypes have been conclusively associated with SQTS (Table 2), but genetic testing was negative in 71% of the reported cases,1 suggesting that other genetic mutations remain to be identified.
Association between genotype and phenotype in short QT syndrome.
| Type | Gene involved | Chromosome locus |
| SQTS1 | KCNH2 – potassium voltage-gated channel, subfamily H (eag-related), member 2 | 7q36.1 |
| SQTS2 | KCNQ1 – potassium voltage-gated channel, KQT-like subfamily, member 1 | 11p15.5-p15.4 |
| SQTS3 | KCNJ2 – potassium inwardly-rectifying channel, subfamily J, member 2 | 17q24.3 |
| SQTS4 | CACNA1C – calcium channel, voltage-dependent, L type, alpha 1C subunit | 12p13.3 |
| SQTS5 | CACNB2 – calcium channel, voltage-dependent, beta-2 subunit | 10p12.33-p12.31 |
| SQTS6 | CACNA2D1 – calcium channel, voltage-dependent, alpha-2/delta subunit 1 | 7q21.11 |
Although genetic testing has so far shown a low diagnostic yield, a hereditary component was found in 71.9% of index cases in the study by Gollob et al.2 However, application of a diagnostic score to family members may underestimate the prevalence of SQTS due to the incomplete penetrance of inherited cardiac electrical disorders. ECG screening of family members appears to be a practical and cost-effective tool.
The ideal long-term management of this rare clinical entity has yet to be established. In theory, pharmacological therapy with a QT-prolonging drug should be effective in correcting the short QT interval; however, most QT-prolonging drugs, such as flecainide or sotalol, are ineffective in these patients.9 Recent data show that most QT-prolonging drugs have the highest affinity for the inactivated state of the potassium channel Kv11.1.10 Curiously, in SQTS, Kv11.1 inactivation is usually abolished, conferring relative resistance to the action of these drugs. An exception is quinidine, which binds with equal affinity to both the activated and inactivated states of Kv11.1, and has proved effective in prolonging the QT interval up to normal in these patients.9,7 A recent report has shown that the arrhythmic event rate in SQTS patients taking hydroquinidine during long-term follow-up was lower than if no drug was used (0.0% vs. 4.9%/year), although this difference was not statistically significant, probably owing to the small sample size.11 Thus, quinidine may be a useful treatment for selected patients with SQTS, such as those with contraindication to or refusing ICD implantation.
Current guidelines2 recommend ICD implantation for patients with genetic arrhythmia syndromes (including SQTS) and prior cardiac arrest or syncope, and also for those with a family history of sudden cardiac death and induction of ventricular tachyarrhythmias, as they are considered to be at high risk for recurrent malignant arrhythmic events. Implantation for primary prevention may be considered in patients classified as high probability on the basis of their Gollob score.1
In our patient, no arrhythmic events or relevant symptoms were recorded during a 14-month follow-up. This is in agreement with the reported low incidence of events in a cohort of SQTS patients,13 with an event rate of 3.3% per year. Specifically, in the subset of patients presenting with a history of syncope, only one event was observed, in a patient with a hERG mutation (SQTS1) who received an appropriate shock for ventricular fibrillation.
Among the causes of sudden cardiac death in young people, SQTS is a relatively recent entity. The recognition of this rare syndrome has allowed reclassification of individuals who would previously have been classified as having idiopathic ventricular fibrillation, as is the case with our patient. Any suspected cases of SQTS should be referred to specialized centers where they can be offered a multidisciplinary approach and disease-specific therapeutic interventions, such as quinidine or ICD implantation, according to SQTS risk stratification. In the future, further advances in our knowledge of inherited channelopathies will hopefully help in the early recognition and prevention of arrhythmic events in this high-risk population.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
