


Isolated left ventricular noncompaction is a rare congenital cardiomyopathy, characterized morphologically by a dilated left ventricle, prominent trabeculations and deep intertrabecular recesses in the ventricular myocardium, with no other structural heart disease. It is thought to be secondary to an arrest of normal myocardial compaction during fetal life. Clinically, the disease presents with heart failure, embolic events, arrhythmias or sudden death. Current diagnostic criteria are based on clinical and imaging data and two-dimensional and color Doppler echocardiography is the first-line exam. There is no specific therapy and treatment is aimed at associated comorbidities. Cases refractory to medical therapy may require heart transplantation.
The authors describe a case of severe and refractory heart failure, which was the initial presentation of isolated left ventricular noncompaction in a previously healthy male child, who underwent successful heart transplantation.
A não compactação isolada do miocárdio ventricular é uma causa rara de miocardiopatia congénita primária, caracterizada morfologicamente por dilatação do ventrículo esquerdo, padrão multitrabecular proeminente e recessos profundos intertrabeculares no miocárdio ventricular, na ausência de outras cardiopatias estruturais. Parece resultar da cessação intrauterina do processo de compactação miocárdica durante a embriogénese. Clinicamente, apresenta-se com sintomas ou manifestações de insuficiência cardíaca, eventos cardioembólicos, disritmias ou morte súbita. Os critérios de diagnóstico baseiam-se em achados clínicos e imagiológicos, sendo o ecocardiograma bidimensional e Doppler o meio complementar de diagnóstico de eleição. Não existe tratamento específico para esta entidade, sendo dirigido às comorbilidades associadas. O transplante cardíaco poderá ser uma possibilidade terapêutica em casos refratários ao tratamento médico.
Os autores descrevem um caso clínico de insuficiência cardíaca refratária como forma de apresentação inaugural de não compactação do miocárdio ventricular numa criança do sexo masculino previamente saudável, submetida com sucesso a transplantação cardíaca.
Isolated left ventricular noncompaction (LVNC) or myocardial noncompaction is a rare congenital cardiomyopathy.1,2 It is thought to be secondary to an arrest of normal myocardial compaction during fetal life and is characterized morphologically by left ventricular (LV) dilatation, prominent myocardial trabeculations and deep intertrabecular recesses, giving the myocardium a spongy appearance.3,9
It has been described as an isolated finding or more often in association with other primary heart disease, particularly cyanotic congenital heart defects.9,10
The natural history of LVNC is varied; patients may remain asymptomatic or progress to congestive heart failure (HF). At pediatric ages, progression to LV systolic and diastolic dysfunction is almost inevitable, occurring in around 90% of affected children. The associated complications, including systemic thromboembolism and arrhythmias, which are linked to high morbidity and mortality, as well as sudden death, are mainly seen in adulthood.3,11
The authors describe a case of this rare disease, which can be difficult to recognize, in a male child with isolated LVNC who underwent successful heart transplantation.
Case reportA 10-year-old boy, white, previously healthy, was admitted to the emergency department due to cough, repeated vomiting and abdominal pain. He reported fatigue with moderate to vigorous exertion (NYHA functional class II) over the previous month. His parents were non-consanguineous, but there was a family history of unexplained sudden death (mother and uncle, grandfather and great-aunt on the mother's side).
At admission, the patient was apyretic, normotensive (blood pressure 100/66 mmHg), tachycardic (heart rate 150 bpm) and tachypneic (respiratory rate 37 cpm). His skin and mucous membranes were pale and there was a grade II/VI systolic murmur over the left sternal border on cardiac auscultation, and hepatomegaly with a palpable liver edge around 3 cm from the right costal margin. No dysmorphic facial features or neuromuscular abnormalities were observed.
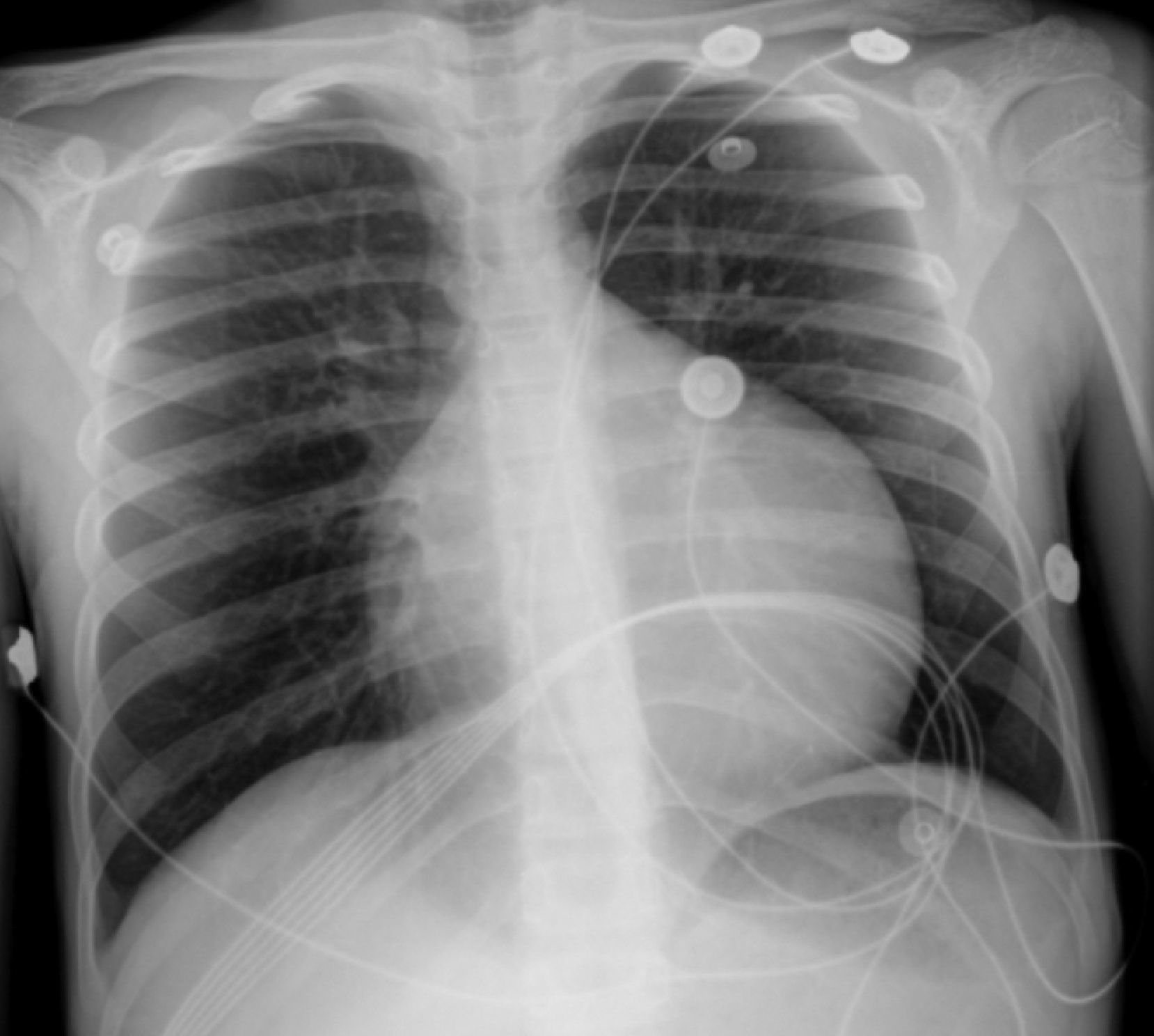
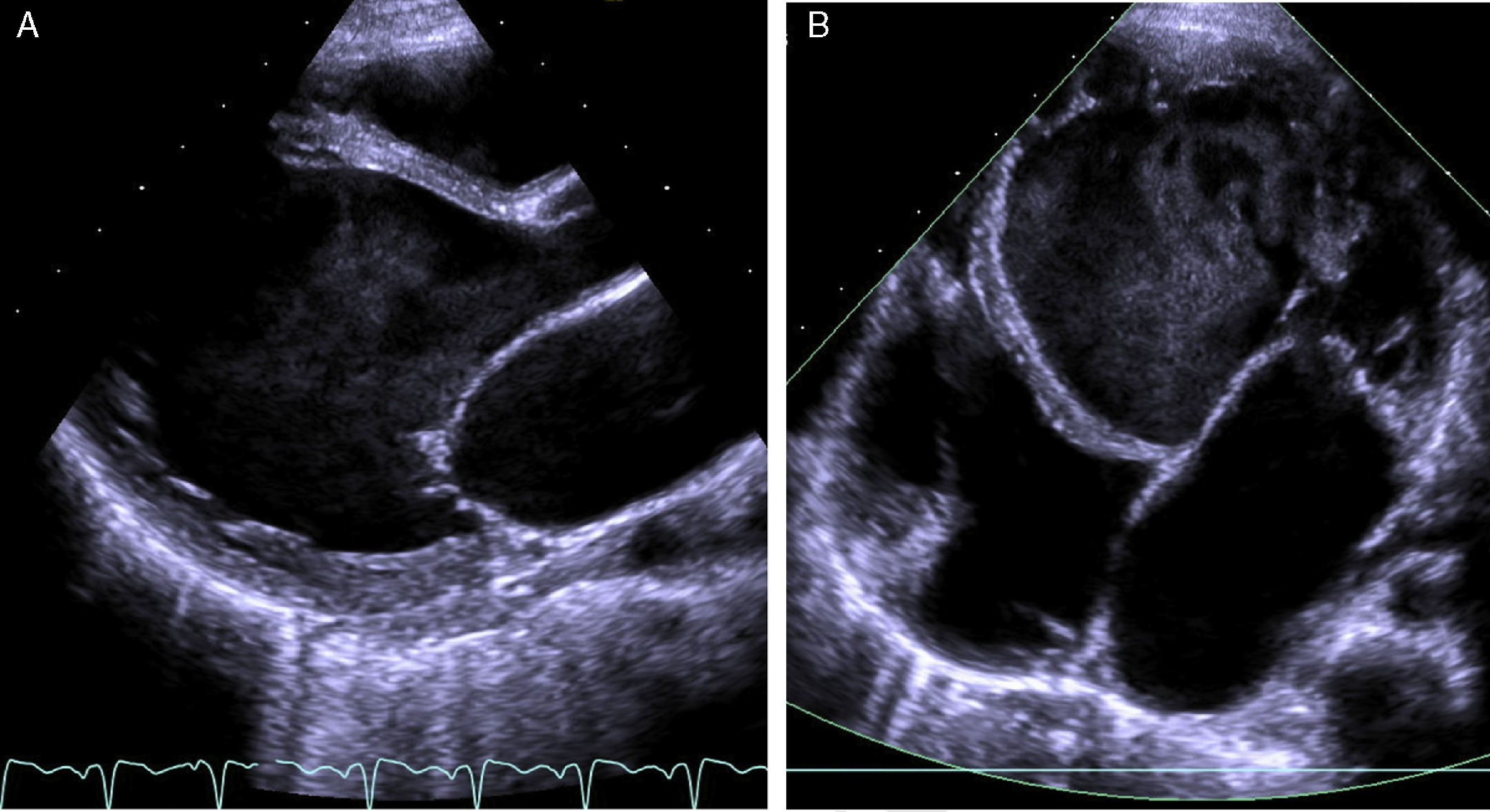
The electrocardiogram (ECG) showed narrow-complex tachycardia, with heart rate between 150 and 180 bpm, that did not respond to administration of adenosine, consistent with ventricular tachycardia. The chest X-ray revealed cardiomegaly (cardiothoracic index >0.6, Figure 1) and laboratory tests showed elevated transaminases (GOT 94 UI/l, GPT 72 UI/l) and BNP (742 pg/ml) and hyponatremia (133 mEq/l). Markers of myocardial necrosis were negative. Transthoracic echocardiography (Figure 2A and B) showed LV dilatation and severe systolic dysfunction (end-diastolic diameter 65 mm, z-score +7.8; end-systolic diameter 60 mm, z-score +12.3; ejection fraction 20%; and fractional shortening 12%). Prominent trabeculations and deep intertrabecular recesses filled with blood were visualized in the LV lateral wall and apical region; the ratio between the thickness of the noncompacted and compacted layers was >2, measured in short-axis parasternal view at end-systole, in accordance with the criteria of Jenni et al.16
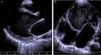
Cardiac magnetic resonance imaging (CMRI) confirmed the diagnosis of LVNC, revealing endomyocardial hypertrabeculation of the LV lateral wall and apical region, global hypokinesia and no late gadolinium enhancement (Figure 3A and B). The ratio between the thickness of the noncompacted and compacted layers was >2.3 measured at end-diastole, in accordance with the criteria of Petersen et al.13
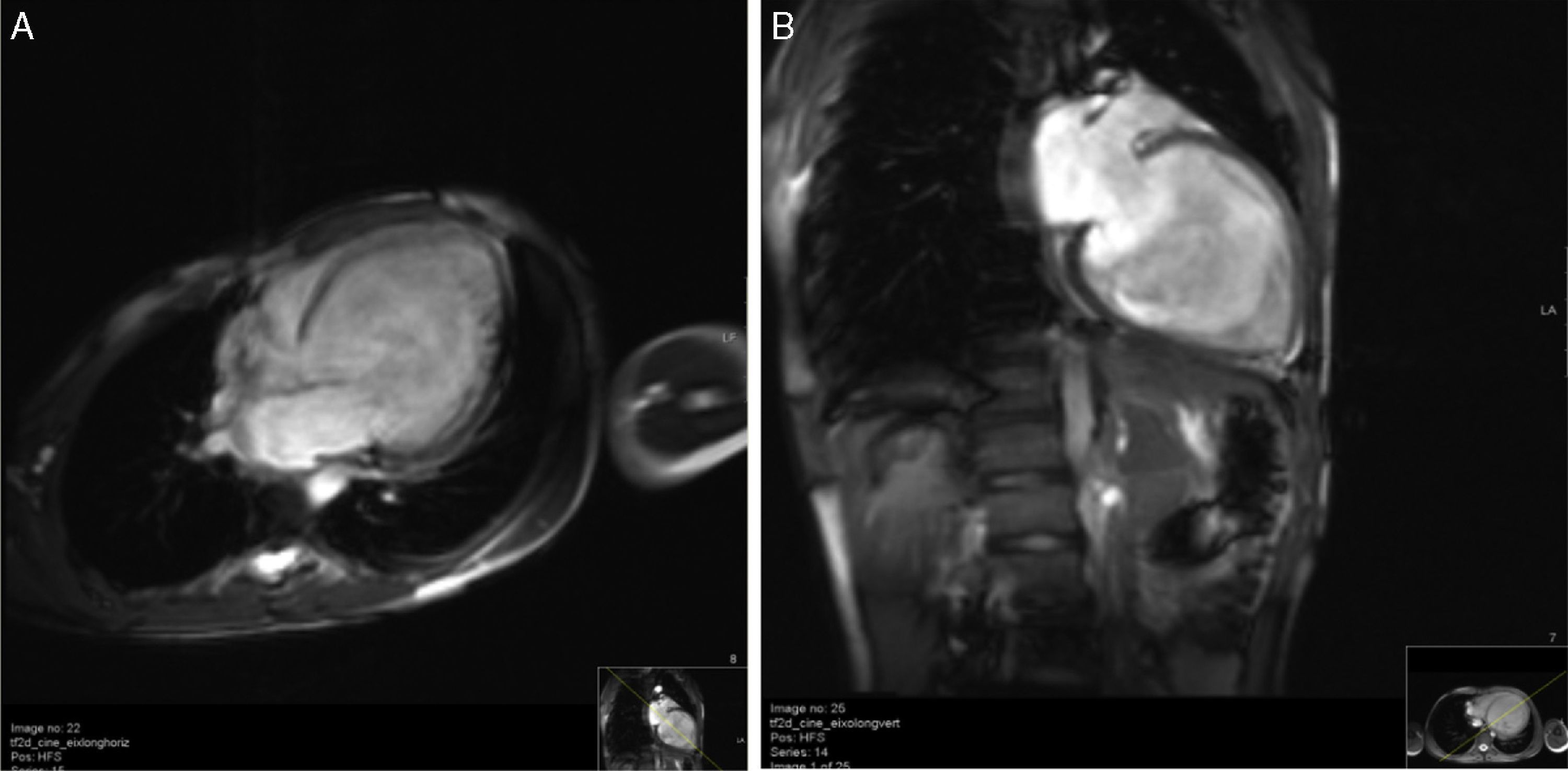
In the light of the imaging diagnosis, genetic study was performed to screen for mutations associated with dilated cardiomyopathy, LVNC or neuromuscular or conduction disorders, but none were identified. Metabolic study was negative. Echocardiographic screening of first-degree relatives was positive, dilated cardiomyopathy being detected in the patient's brother.
Cardiac catheterization showed mean pulmonary artery pressure of 28 mmHg, pulmonary wedge pressure of 18 mmHg, and pulmonary vascular resistance of 3.5 Wood units.
HF therapy was begun with diuretics, angiotensin-converting enzyme inhibitors and antiplatelet agents.
After admission, the patient's clinical condition worsened, prompting a two-day stay in the intensive care unit with need for hemodynamic support, initially with milrinone and subsequently with levosimendan. Anticoagulation with enoxaparin was begun due to an apparent intracardiac thrombus in the right atrium, with no evidence of systemic thromboembolism. During hospitalization, there were short episodes of narrow-complex ventricular tachycardia, without hemodynamic repercussions. Electrophysiological study with programmed ventricular stimulation induced a brief episode of slow, sustained ventricular tachycardia (maximum heart rate 140 bpm). It was decided to begin amiodarone and to place an implantable cardioverter-defibrillator (ICD). The clinical course was favorable, and the patient was discharged on the 36th day with improved clinical condition and laboratory results, but with unchanged imaging features, under anticongestive, anti-arrhythmic and oral anticoagulant therapy.
A month and a half after discharge, he was admitted to the emergency department in cardiogenic shock following cardiopulmonary arrest that required 20 min of resuscitation maneuvers.
Due to the patient's severe ventricular dysfunction, he was placed on the priority list for heart transplantation, which was performed four days later, without complications. The surgical approach adopted was an orthotopic heart transplant using the biatrial technique, reflecting the cardiothoracic surgeon's experience, and the ICD and leads were removed. Inotropic support was discontinued on the second post-operative day, and the patient was extubated on the following day. Immunosuppressive therapy was begun with cyclosporine, azathioprine and prednisone. On the seventh post-operative day, the patient suffered generalized tonic-clonic seizures, followed by blurred vision. The electroencephalogram showed a highly abnormal trace, with epileptiform activity, and therapy was begun with phenytoin. Head computed tomography (CT) showed subacute bilateral ischemic lesions in the cortical (frontal, parietal, occipital and temporal) and subcortical (cerebellar) regions. Subsequent neurological improvement and total recovery of the patient's vision led to a probable diagnosis of reversible posterior encephalopathy syndrome.
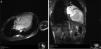
Anatomopathological study of the removed heart showed marked trabeculation affecting around two-thirds of the thickness of the LV inner wall, with recesses communicating with the ventricular cavity and evidence of thrombi and focal fibrosis, which confirmed the diagnosis of LVNC (Figure 4). The patient was discharged on the 21st post-operative day, stable in hemodynamic and imaging terms, under anticonvulsant and immunosuppressive therapy and prophylactic therapy against opportunistic infections. Clinical follow-up, aided by complementary exams, hemodynamic study and endomyocardial biopsy, revealed grade 2R (moderate acute rejection), and the initial immunosuppressive regimen was changed to tacrolimus and mycophenolate mofetil, which has been maintained to date.
Discussion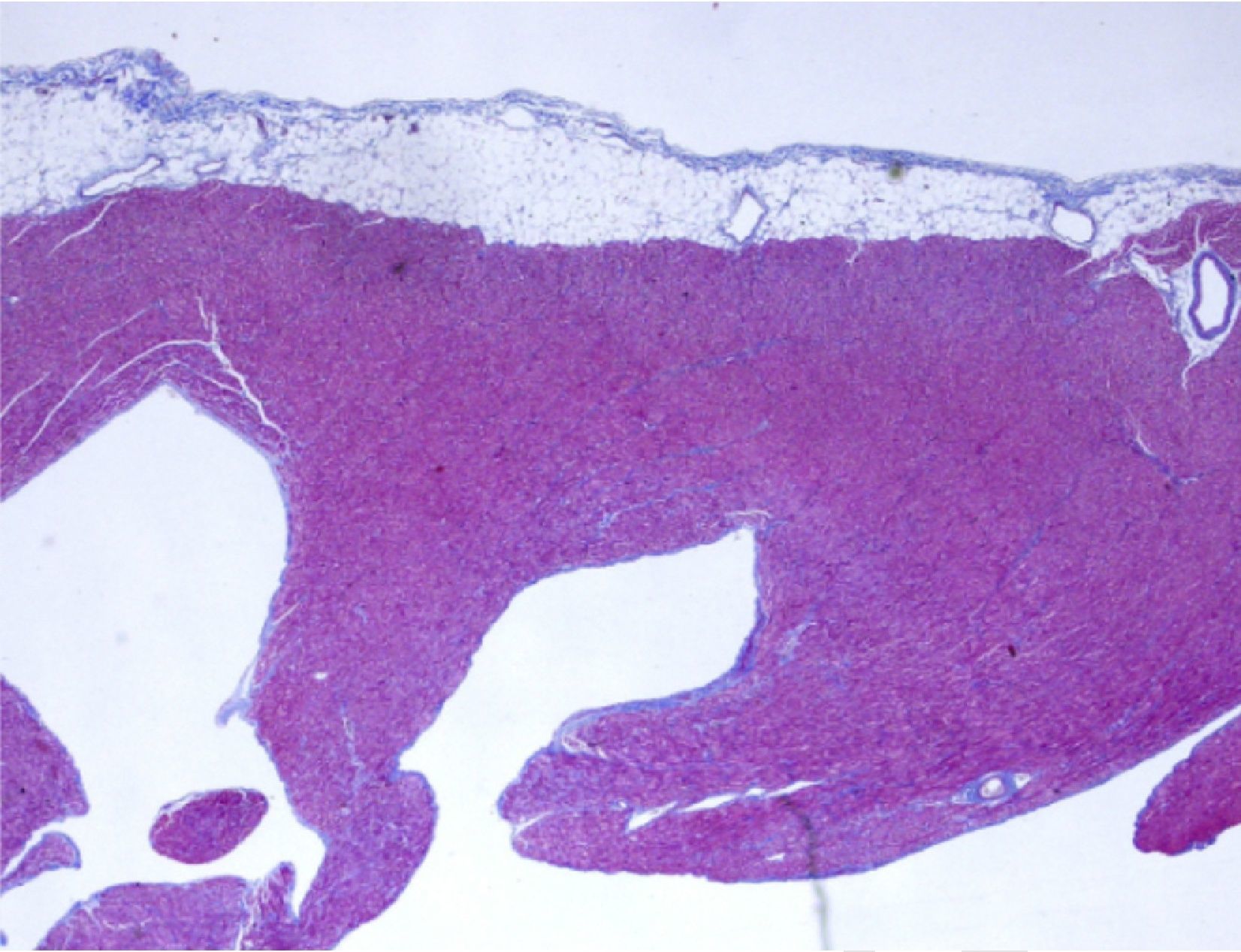
Isolated LVNC is a rare disease that is difficult to recognize and of uncertain etiology. Its incidence in the pediatric population is 0.01% according to various echocardiographic studies.3,8,9,13 It appears to be more common in males (56–82% of cases).6,8,11,12 Age at diagnosis varies greatly, from the fetus4,20 to the elderly. At pediatric ages, it is more frequent in early infancy.9
Isolated LVNC was first described by Chin et al.6 in 1990 and has since been widely studied; it was classified as a congenital cardiomyopathy in 2006 by the American Heart Association.1
In normal embryonic development, the myocardium consists of a spongy meshwork of fibers and intertrabecular spaces.3,12,14,15 During endomyocardial morphogenesis, which occurs between the 5th and 8th week of intrauterine development, the future ventricular myocardium becomes compacted, primitive sinusoids are transformed into blood capillaries and the coronary circulation develops.3,16,17
It is generally accepted that isolated LVNC results from an arrest in the process of myocardial compaction in the absence of other structural heart disease or primary hemodynamic or functional abnormalities.3,6 It is characterized by multiple prominent trabeculations and intertrabecular recesses that penetrate deep into the muscle wall of the affected areas and communicate only with the ventricular cavity.11,18 It is distinguished from the more common non-isolated form associated with other primary heart disease such as ventricular septal defect, obstructive lesions of the left and right outflow tracts, complex cyanotic heart disease and coronary artery anomalies,9,10,12,15,20 in which the pattern of noncompaction is histologically different and may in fact be secondary to these malformations.8 It should be noted that ventricular hypertrabeculation can be seen in healthy hearts,16 as well as in dilated, hypertrophic and restrictive cardiomyopathy,5,9,13 and some of the characteristic changes can be found in other entities, including apical hypertrophic cardiomyopathy, right ventricular arrhythmogenic dysplasia, endocardial fibroelastosis, cardiac metastases and intraventricular thrombi.3,11,14 Associations have also been reported with neuromuscular disease, metabolic disorders and facial dysmorphisms.6,11
Isolated LVNC can present in sporadic or familial form,9,21 the latter accounting for 20–50% of cases. Various patterns of inheritance have been described, including autosomal dominant and X-linked, a mutation of the G4.5 gene on chromosome Xq28 having been identified in the latter case.21,23
The clinical spectrum is highly varied and nonspecific,24 and manifestations are proportional to the extent of cardiac segments affected.11 Patients may initially be asymptomatic or present HF, arrhythmias, cardioembolic events or even sudden death.3,6,12 In most cases, isolated LVNC eventually progresses to HF and LV systolic and diastolic dysfunction.11 Systolic dysfunction is probably the consequence of decreased oxygen supply due to defects in the coronary microcirculation, while diastolic dysfunction appears to result from fibrosis secondary to ischemia and myocardial disarray.3,14,25 In children, the natural history of isolated LVNC is development of ventricular systolic dysfunction in around 90% of cases, but complications and death are less frequent than in adults.3,11
The most common arrhythmias are atrial fibrillation and ventricular tachycardia, which account for almost half of associated mortality.3,8,12 Other frequent findings in children are paroxysmal supraventricular tachycardia, third-degree atrioventricular block and Wolff-Parkinson-White syndrome.11
Less common are cardioembolic complications such as pulmonary thromboembolism, stroke, transient ischemic attack and mesenteric infarction, which appear to result from a combination of myocardial dysfunction, blood pooling in the sinusoids and arrhythmic events.3,6,8,12
The morphological criteria for a diagnosis of isolated LVNC are based on imaging data, two-dimensional and Doppler echocardiography being the first-line exam.4,6,8,9,16 The most widely used criteria are those established by Jenni et al. in echocardiographic studies of adults, in which a ratio of >2 between the maximal thickness of the endocardial trabeculated layer and that of the epicardial compacted layer, measured in end-systolic short-axis parasternal view, is diagnostic.3,16 Some authors use a ratio of >1.4 in children.9 The other main characteristics of isolated LVNC are the presence of multiple prominent trabeculations and intertrabecular recesses located predominantly in the apical region and the inferior and lateral wall,5,8,16 color Doppler evidence of blood flow from the ventricular cavity to the recesses,3,4,9 and the absence of other cardiac anomalies. Systolic and diastolic dysfunction and diffuse hypokinesia of the entire left ventricle, not only the noncompacted segments, are also observed.8,12,17 The right ventricle appears to be affected in around 40% of cases, but such involvement has been called into question because of the heavily trabeculated nature of the normal right ventricular apex.8,16
Transthoracic echocardiography has certain limitations and when it cannot confirm or exclude a diagnosis of LVNC, other imaging options include transesophageal or contrast echocardiography,3 CT angiography26 and CMRI.13,22
In a recent study on the use of CMRI in LVNC, Petersen et al.13 proposed an end-diastolic ratio of >2.3 between the noncompacted and compacted layers. This technique shows a good correlation with echocardiography but appears to be better at identifying and locating the characteristic pattern, especially in the apical region, which is more difficult to visualize by echocardiography.13,27
In the case presented, the diagnosis was made primarily by echocardiography based on established diagnostic criteria, and was then confirmed by CMRI and anatomopathological study.
Treatment is conservative and directed at the main complications. In patients with HF, it consists of medical therapy with diuretics, angiotensin-converting enzyme inhibitors and beta-blockers.3,9 Arrhythmic events should be thoroughly investigated, and a biventricular pacemaker or ICD considered in cases of ventricular arrhythmias. Antiplatelet therapy is recommended for prevention of systemic embolic events and anticoagulation if there is evidence of thrombi.3,8,9,12
LVNC has a poor prognosis, the most common causes of death being refractory HF, ventricular tachycardia, thromboembolic events and sudden death.8 In previous studies, mortality has exceeded 35% in adults28 and 22% in children,29 depending on the length of follow-up. In refractory or end-stage HF, heart transplantation may be the only option,30,31 as in the case reported.
Given the familial nature of isolated LVNC and its association with neuromuscular disorders, it is essential to perform neurological and echocardiographic assessment of close relatives.
ConclusionThe authors describe a case of dilated cardiomyopathy due to isolated LVNC, a rare indication for pediatric heart transplantation. The diagnosis should be considered in a child with HF or arrhythmic or embolic events, since it may be successfully treated by heart transplantation.
Ethical responsibilitiesProtection of people and animalsThe authors declare that no experiments were performed on humans or animals for this study.
Data confidentialityThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Magalhães M, Costa P, Vaz MT, Pinheiro Torres J, Areias JC. Ventrículo esquerdo não compactado: causa rara de transplante cardíaco. Rev Port Cardiol. 2016;35:61.e1–61.e6.
