

In recent years, the importance of genetic causes of cardiovascular diseases has been increasingly recognized, as the result of significant advances in molecular diagnosis techniques. This growing knowledge has enabled the identification of new phenotypes and the subclassification of clinical syndromes, impacting the therapeutic approach and genetic counseling offered to affected families.
This paper describes the state of the art of genetic testing in the main cardiovascular diseases, aiming to provide a useful tool to help cardiologists and other health professionals involved in the care of individuals with hereditary heart diseases and their families.
Nos últimos anos, tem sido crescente o reconhecimento das causas genéticas das doenças cardiovasculares resultante dos significativos progressos das técnicas laboratoriais. Este conhecimento tem permitido a identificação de «novos» fenótipos e a subclassificação das síndromes clínicas, tendo impacto nas decisões terapêuticas e no aconselhamento genético que é facultado às famílias.
No presente documento descreve-se o «estado da arte» relativamente às principais recomendações para testes genéticos nas doenças cardiovasculares, pretendendo-se providenciar uma ferramenta útil de consulta para Cardiologistas e outros Profissionais envolvidos na prestação nos cuidados de saúde a doentes com cardiopatias hereditárias e respetivas famílias.
In recent years, the importance of genetic causes of cardiovascular diseases has been increasingly recognized, and technical advances have enabled many of the underlying molecular mechanisms to be understood in detail.
The great strides in genetic sequencing, especially next-generation sequencing, have increased the range of molecular diagnosis, but in many cases this has resulted in the detection of genetic variations of as yet uncertain significance, which complicates genetic counseling for patients and their relatives. It is thus essential for cardiologists and other health professionals involved in the care of individuals with hereditary heart diseases and their families to be familiar with the indications, advantages and limitations of genetic tests.
This document describes the main indications for genetic testing in the most important inherited cardiovascular syndromes and diseases and for postmortem genetic testing.
General recommendationsWhenever appropriate, physicians should inform their patients about the mechanisms of the hereditary disease in question and the implications for their relatives, and refer them for a medical genetics consultation.
Genetic counseling should take place both before and after genetic testing, in order to ensure that patients understand the benefits and limitations of the results, and only after patients have provided informed consent. Results should be communicated only to the patient him- or herself.
ChildrenGenetic testing should be requested for children only if it can be of immediate benefit to them and with the informed consent of their parents or guardians. Tests that would reveal a disease that usually has onset in adulthood and for which there is no proven prevention or cure should not be requested. Whenever appropriate, the child should be involved in the process according to his or her degree of autonomy.
Presymptomatic testsPresymptomatic tests are defined as those that can identify a currently asymptomatic individual as a carrier of a genetic variant that is unequivocally associated with a pathological condition. In healthy individuals, presymptomatic tests should only be performed in the context of a medical genetics consultation, following genetic counseling and provision of written informed consent. The results should only be communicated to the patient him- or herself and not to third parties, including physicians not directly involved in the testing process, without written authorization.
Genetic testing in Portugal is regulated by Decree-Law 12/2005, the relevant parts of which are presented in Supplement 1.
The main genetic tests used in clinical practice, the genes referred to, and abbreviations used in this document, are listed in Supplements 2-4, respectively.
Classification of genetic variantsGenetic variants should be classified as one of five categories: pathogenic, likely pathogenic, of uncertain significance, likely benign, or benign.1 Only pathogenic or likely pathogenic variants should be used to direct specialized follow-up and to prompt genetic study of an at-risk carrier and asymptomatic relatives.
Classes of recommendation and levels of evidenceMost of the information available is derived from registries and non-randomized studies and is thus level of evidence C. The classes of recommendation and levels of evidence used in this document are presented in Supplement 5.
Genetic testing in relativesWhen a pathogenic or likely pathogenic genetic variant is identified in an index case, it is a class I recommendation,2,3 as with all hereditary heart diseases, to perform genetic testing in relatives, together with genetic counseling both before and after testing.
ChannelopathiesChannelopathies are primary electrical diseases of the heart that are not accompanied by macroscopic or histopathological abnormalities detectable by conventional means, since the functional and structural alterations occur at the molecular level, in the cell membrane.4 They constitute a varied group of diseases in which pathogenic variants in genes that code for ion channels cause changes in the ion currents involved in the action potential of cardiac cells, leading to potentially fatal arrhythmias.5,6
J-wave syndromesJ-wave syndromes are those in which an elevated J wave (appearing as a J point) on the electrocardiogram (ECG) is associated with increased risk of ventricular arrhythmias.7 Among the best-known of these syndromes are Brugada syndrome (BrS) and early repolarization syndrome (ERS), which are associated with the development of polymorphic ventricular tachycardia (VT), ventricular fibrillation (VF) and sudden death (SD). J-wave/J-point abnormalities occur in different leads of the ECG in BrS (right precordial leads) and ERS (mainly in the inferior and lateral leads).7,8
Brugada syndromeClinical diagnosisDiagnosis of BrS is made in the presence of a type 1 pattern on the ECG (coved-type ST-segment elevation of ≥2 mm in one or more of the right precordial leads, V1 and/or V2, in the second, third or fourth intercostal space), spontaneously or following provocation with a sodium channel blocker such as flecainide or ajmaline.8–10
To avoid overdiagnosis, when a type 1 pattern has been documented on the ECG only after pharmacological provocation, at least one of the following criteria should also be fulfilled for a diagnosis of BrS: VF or polymorphic VT; syncope of probable arrhythmic cause; family history of SD before the age of 45 years with negative autopsy; type 1 pattern in relatives; nocturnal agonal respiration; or inducibility of VT/VF.8 Table S1 in Supplement 6 presents a diagnostic score for BrS.8
Genetic diagnosisBrS is associated with genetic variants in multiple genes, most of which code for sodium channels (SCN5A in 11-28% of probands, SCN10A in 5-17%) or calcium channels (CACNA1C in 7% and CACNB2b in 5%).8
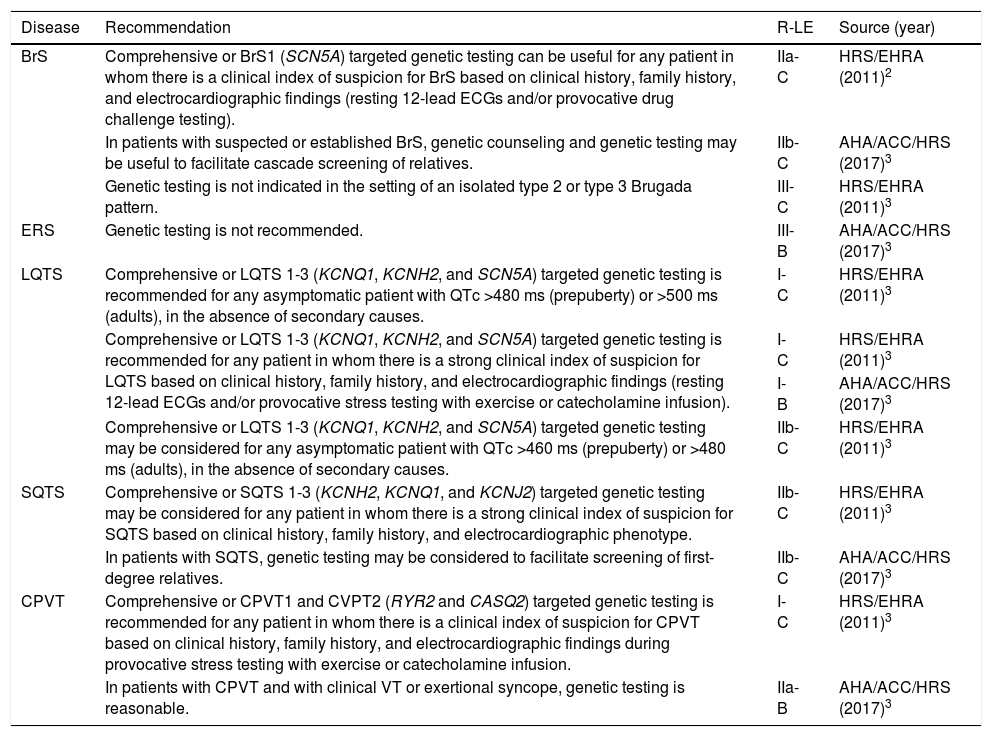
Genetic testing is not necessary for the diagnosis, but can be useful in cases of doubtful phenotypes and in those with established BrS (class IIb3 or IIa2 recommendation), particularly to facilitate familial genetic screening (class IIb3) (Table 1).
Recommendations for genetic testing in channelopathies.
| Disease | Recommendation | R-LE | Source (year) |
|---|---|---|---|
| BrS | Comprehensive or BrS1 (SCN5A) targeted genetic testing can be useful for any patient in whom there is a clinical index of suspicion for BrS based on clinical history, family history, and electrocardiographic findings (resting 12-lead ECGs and/or provocative drug challenge testing). | IIa-C | HRS/EHRA (2011)2 |
| In patients with suspected or established BrS, genetic counseling and genetic testing may be useful to facilitate cascade screening of relatives. | IIb-C | AHA/ACC/HRS (2017)3 | |
| Genetic testing is not indicated in the setting of an isolated type 2 or type 3 Brugada pattern. | III-C | HRS/EHRA (2011)3 | |
| ERS | Genetic testing is not recommended. | III-B | AHA/ACC/HRS (2017)3 |
| LQTS | Comprehensive or LQTS 1-3 (KCNQ1, KCNH2, and SCN5A) targeted genetic testing is recommended for any asymptomatic patient with QTc >480 ms (prepuberty) or >500 ms (adults), in the absence of secondary causes. | I-C | HRS/EHRA (2011)3 |
| Comprehensive or LQTS 1-3 (KCNQ1, KCNH2, and SCN5A) targeted genetic testing is recommended for any patient in whom there is a strong clinical index of suspicion for LQTS based on clinical history, family history, and electrocardiographic findings (resting 12-lead ECGs and/or provocative stress testing with exercise or catecholamine infusion). | I-C | HRS/EHRA (2011)3 | |
| I-B | AHA/ACC/HRS (2017)3 | ||
| Comprehensive or LQTS 1-3 (KCNQ1, KCNH2, and SCN5A) targeted genetic testing may be considered for any asymptomatic patient with QTc >460 ms (prepuberty) or >480 ms (adults), in the absence of secondary causes. | IIb-C | HRS/EHRA (2011)3 | |
| SQTS | Comprehensive or SQTS 1-3 (KCNH2, KCNQ1, and KCNJ2) targeted genetic testing may be considered for any patient in whom there is a strong clinical index of suspicion for SQTS based on clinical history, family history, and electrocardiographic phenotype. | IIb-C | HRS/EHRA (2011)3 |
| In patients with SQTS, genetic testing may be considered to facilitate screening of first-degree relatives. | IIb-C | AHA/ACC/HRS (2017)3 | |
| CPVT | Comprehensive or CPVT1 and CVPT2 (RYR2 and CASQ2) targeted genetic testing is recommended for any patient in whom there is a clinical index of suspicion for CPVT based on clinical history, family history, and electrocardiographic findings during provocative stress testing with exercise or catecholamine infusion. | I-C | HRS/EHRA (2011)3 |
| In patients with CPVT and with clinical VT or exertional syncope, genetic testing is reasonable. | IIa-B | AHA/ACC/HRS (2017)3 |
ACC: American College of Cardiology; AHA: American Heart Association; BrS: Brugada syndrome; CPVT: catecholaminergic polymorphic ventricular tachycardia; ECG: electrocardiogram; EHRA: European Heart Rhythm Association; ERS: early repolarization syndrome; HRS: Heart Rhythm Society; LE: level of evidence; LSQTS: long QT syndrome; QTc: corrected QT interval; R: class of recommendation; SQTS: short QT syndrome; VT: ventricular tachycardia.
Diagnosis of ERS is based on a pattern of early repolarization in the inferior and/or lateral leads and a history of aborted SD, documented VF or polymorphic VT.8 ER is recognized if (1) there is an end-QRS notch (J wave) or slur on the downslope of a prominent R wave with and without ST-segment elevation; (2) the peak of the notch or J wave (Jp) ≥0.1 mV in ≥2 contiguous leads of the 12-lead ECG, excluding leads V1-V3; and (3) QRS duration (measured in leads in which a notch or slur is absent) of <120 ms.11 Table S2 in Supplement 6 presents a diagnostic score for ERS.8
Genetic diagnosisERS has so far been associated with genetic variants in seven genes, mainly coding for calcium channels (CACNA1C, CACNB2b and CACNA2D1), but their etiological role is questionable8 and so genetic testing is not recommended3 (Table 1).
QT syndromesLong QT syndromeClinical diagnosisThe diagnosis of long QT syndrome (LQTS) is based on measurement of the QT interval after excluding secondary causes of QT prolongation such as drugs or electrolyte imbalances.10 To help with diagnosis, a score has been created12 that as well as corrected QT interval (QTc) also considers other electrocardiographic abnormalities, symptoms, and family history (Table S3, Supplement 6). The diagnosis is now established with either QTc ≥480 ms in repeated ECGs (class I) or LQTS risk score >3 (class I), or in the presence of a confirmed pathogenic LQTS mutation, irrespective of the QT duration (class I); it should be considered in the presence of QTc ≥460 ms in repeated 12-lead ECGs in patients with unexplained syncope.9
Genetic diagnosisLQTS is associated with documented genetic variants in at least 15 genes.13 Genetic testing identifies pathogenic variants in around 75% of cases, and three genes are responsible for about 90% of positive tests: KCNQ1, KCNH2 and SCN5A (associated with types 1, 2 and 3 LQTS, respectively.9 Genetic testing is of particular value in these cases, since the three genotypes are associated with different degrees of SD risk, especially in association with gender and QTc duration (the risk is highest in women with type 2 LQTS and in men with type 3 LQTS and QTc >500 ms6,14), and since the triggers for arrhythmic events also differ (physical exertion, particularly swimming, in type 1; sudden loud noises in type 2; and rest or sleep in type 3).6,15 Around 20-25% of patients with genetically confirmed LQTS have a normal QTc.10,16
Molecular study may be aimed at a specific gene or may be guided by the ECG, syncope triggers or the characteristics of associated syndromes. In patients with congenital deafness, cardiac malformations, cognitive deficit, autism spectrum disorder and/or dysmorphisms, diagnostic hypotheses should include Jervell and Lange-Nielson, Timothy, or Andersen-Tawil syndromes. Alternatively the use of gene panels that screen multiple genes associated with LQTS can help diagnose rarer forms of the disease.17
Jervell and Lange-Nielson syndrome is an autosomal recessive disorder, more rarely compound heterozygous, involving the genes KCNQ1 or KCNE1, in which long QTc, usually >500 ms, is associated with congenital profound bilateral sensorineural hearing loss. It usually manifests as syncope following sympathetic activation.6,18 Timothy syndrome (LQTS type 8) is associated with pathogenic variants in CACNA1C and is characterized by atrioventricular conduction disturbances, tachyarrhythmias, congenital heart defects, hand/foot and facial dysmorphism, neurodevelopmental problems and autism spectrum disorder.6,19 Andersen-Tawil syndrome (LQTS type 7), associated with pathogenic variants of KCNJ2, besides prolonged QTc is characterized by the presence of U waves, hypokalemic periodic paralysis, facial dysmorphism and mild neurocognitive deficits, and regularly presents with palpitations, syncope, or periodic paralysis triggered by prolonged rest or rest following exertion.6,20
Genetic testing and genetic counseling are recommended in all individuals diagnosed as having or with a strong clinical suspicion of LQTS (class I),2,3 and may be considered in asymptomatic individuals with QTc >460 ms (prepuberty) or >480 ms (adults), in the absence of secondary causes (class IIb)2 (Table 1).
Short QT syndromeClinical diagnosisSQTS is diagnosed in the presence of QTc ≤340 ms (class I), and should be considered (class IIa) in the presence of QTc ≤360 ms and one or more of the following: a confirmed pathogenic mutation; a family history of SQTS; a family history of SD at age <40 years; or survival from a VT/VF episode in the absence of structural heart disease.9
Genetic diagnosisSQTS is associated with variants in three genes coding for potassium channels (KCNH2, KNCQ1 and KCNJ2), which are also associated with LQTS, although with the opposite effect in terms of functional alterations.6,10
Genetic testing may be considered in individuals with SQTC (class IIb)2,3 to facilitate screening of first-degree relatives.3 Unlike in LQTS, genetic testing has no prognostic value in SQTS.
Catecholaminergic polymorphic ventricular tachycardiaClinical diagnosisCatecholaminergic polymorphic ventricular tachycardia (CPVT) is a hereditary arrhythmogenic syndrome that typically manifests with adrenergically mediated syncope or SD secondary to VT.21 Diagnosis is established by in the presence of a structurally normal heart, normal ECG and exercise- or emotion-induced bidirectional or polymorphic VT (class I) or in patients who are carriers of pathogenic mutation(s) in the genes RyR2 or CASQ2 (class I).9
Genetic diagnosisGenetic testing is recommended in individuals with CPVT (class IIa3/class I2) (Table 1).
CardiomyopathiesThe cardiomyopathies are diseases in which the heart muscle is structurally and functionally abnormal, in the absence of coronary artery disease, hypertension, valvular disease or congenital heart disease sufficient to cause the observed myocardial abnormality.22
Molecular study is nowadays an integral part of the assessment and management of patients with cardiomyopathies and their families, and is also used for diagnosis of familial forms.23,24
Hypertrophic cardiomyopathyClinical diagnosisHypertrophic cardiomyopathy (HCM) is defined by the presence of inappropriate left ventricular hypertrophy disproportionate to loading conditions or other cardiac or systemic factors sufficient to cause the observed anomaly. The diagnostic criterion is a maximum wall thickness of ≥15 mm in any left ventricular myocardial segment. In first-degree relatives, diagnosis is established by an otherwise unexplained wall thickness of ≥13 mm in any segment.25
Genetic diagnosisGenetic testing (targeted or multiple gene panels) is recommended in patients with a clinical diagnosis of HCM (class I,2,25 class IIa,3,26 level A23), particularly when screening of family members is anticipated.25,26 Molecular diagnosis is also recommended when the clinical presentation suggests a particular genetic cause that is non-sarcomeric (class I).26
In patients fulfilling diagnostic criteria for HCM, genetic testing is positive in 30-60% of cases,25,27–29 higher in familial disease and lower in older patients and in those with non-standard clinical manifestations, including late onset, less severe hypertrophy, concentric hypertrophy or sigmoid septum, and absence of adverse events.25,28,29
Genetic testing in patients with a borderline diagnosis of HCM (left ventricular wall thickness 12-13 mm in adults; left ventricular hypertrophy in the presence of hypertension, athletic training, valve disease) should be performed only after detailed assessment by specialist teams (class IIa),25 since the result may also be equivocal: a negative result does not rule out the diagnosis and variants of uncertain significance are hard to interpret25 (Table 2).
Recommendations for genetic testing in cardiomyopathies.
| Disease | Recommendation | R-LE | Source (year) |
|---|---|---|---|
| HCM | Genetic testing is recommended in patients fulfilling diagnostic criteria for HCM to confirm the diagnosis. | I-B | ESC (2014)25 |
| Comprehensive or targeted (MYBPC3, MYH7, TNNI3, TNNT2, TPM1) genetic testing is recommended for any patient with a clinical diagnosis of HCM based on clinical history, family history, and electrocardiographic/ echocardiographic findings. | I-C | HRS/EHRA (2011)2 | |
| Genetic testing for HCM and other genetic causes of unexplained cardiac hypertrophy is recommended in patients with an atypical clinical presentation of HCM or when another genetic condition is suspected to be the cause. | I-B | ACCF/AHA (2011)26 | |
| Genetic testing is reasonable in individuals with confirmed or clinically suspected diagnosis of HCM to facilitate the identification of first-degree family members at risk for developing HCM. | IIa-B | HRS/EHRA (2011)2 | |
| ACCF/AHA (2011)26 | |||
| Genetic testing in patients with a borderline diagnosis of HCM should be performed only after detailed assessment by specialist teams. | IIa-C | ESC (2014)25 | |
| Genetic testing should be considered in the most clearly affected person in a family to facilitate family screening and management. | A | HFSA (2018)23 | |
| DCM | Comprehensive or targeted (LMNA and SCN5A) genetic testing is recommended for patients diagnosed with DCM and significant cardiac conduction disease and/or a family history of premature unexpected SD. | I-C | HRS/EHRA (2011)2 |
| Genetic testing is recommended in the presence of a familial form of DCM or in sporadic DCM with clinical clues suggestive of a particular/rare genetic disease. | I | ESC (2016)24 | |
| Genetic testing can be useful for patients with familial DCM to confirm the diagnosis, to recognize those who are at highest risk of arrhythmia and syndromic features. | IIa-C | HRS/EHRA (2011)2 | |
| Genetic testing can be useful for patients with familial DCM to confirm the diagnosis, to facilitate cascade screening within the family, and to help with family planning. | mod-A | AHA (2016)31 | |
| IIa-C | HRS/EHRA (2011)2 | ||
| In patients with familial or idiopathic DCM, genetic testing can be useful in conjunction with genetic counseling. | mod-B | AHA (2016)31 | |
| Genetic testing should be considered in the most clearly affected person in a family to facilitate family screening and management. | A | HFSA (2018)23 | |
| ARVC | Comprehensive or targeted (DSC2, DSG2, DSP, JUP, PKP2, and TMEM43): | HRS/EHRA (2011)2 | |
| - can be useful for patients satisfying the diagnostic criteria for ARVC of the 2010 Task Force32 | IIa-C | ||
| - may be considered for patients with possible ARVC (1 major or 2 minor criteria of the 2010 Task Force32 | IIb-C | ||
| - is not recommended for patients with only a single minor criterion of the 2010 Task Force.32 | III-C | ||
| In patients with clinically diagnosed or suspected ARVC, genetic testing can be useful for diagnosis and for gene-specific targeted family screening. | IIa-B | AHA/ACC/HRS (2017)3 | |
| Genetic testing should be considered in the most clearly affected person in a family to facilitate family screening and management. | A | HFSA (2018)23 | |
| RCM | Genetic testing may be considered for patients with a clinical index of suspicion of HCM based on clinical history, family history, and electrocardiographic/ echocardiographic findings. | IIb-C | HRS/EHRA (2011)2 |
| Genetic testing should be considered in the most clearly affected person in a family to facilitate family screening and management. | B | HFSA (2018)23 | |
| LVNC | Genetic testing may be considered for patients with a clinical diagnosis of LVNC based on clinical history, family history, and electrocardiographic/echocardiographic findings. | IIa-C | HRS/EHRA (2011)2 |
ACC: American College of Cardiology; AHA: American Heart Association; ARVC: arrhythmogenic right ventricular cardiomyopathy; DCM:dilated cardiomyopathy; EHRA: European Heart Rhythm Association; HCM: hypertrophic cardiomyopathy; HFSA: Heart Failure Society of America; HRS: Heart Rhythm Society; LE: level of evidence; LVNC: left ventricular non-compaction; mod: moderate level of evidence; R: class of recommendation; RCM: restrictive cardiomyopathy; SD: sudden death.
Metabolic and other hereditary diseases account for a small, though important, proportion of patients with an HCM genotype. Conditions that more commonly cause HCM include Anderson-Fabry disease, Danon disease, amyloidosis and HCM caused by variants in the PRKAG2 gene. Differential diagnosis is crucial, since these diseases have very different natural history and prognosis, and may require different therapeutic approaches. Supplement 7 lists cardiac and extracardiac manifestations that can guide molecular diagnosis.
Dilated cardiomyopathyClinical diagnosisDilated cardiomyopathy (DCM) is defined by the presence of left ventricular or biventricular dilatation and systolic dysfunction in the absence of abnormal loading conditions or coronary artery disease sufficient to cause the observed degree of dysfunction. It is considered phenotypically related to hypokinetic non-dilated cardiomyopathy (HNDC), which is defined by the presence of left ventricular or biventricular systolic dysfunction (defined as left ventricular ejection fraction <45%) without dilatation.24
As molecular diagnostic techniques have improved, 25-50% of cases of DCM previously thought to be idiopathic have been found to have a genetic basis, mainly with autosomal dominant transmission.24,30,31 A comprehensive investigation of family history in index cases, covering at least three generations, is thus essential. In the absence of conclusive molecular genetic information in a family, DCM or HNDC is considered to be familial if two or more family members are affected or in the presence of an index patient fulfilling diagnostic criteria for DCM/HNDC and a first-degree relative with autopsy-proven DCM and SD by the age of 50 years.24
However, the lack of a family history does not exclude a genetic cause, and the latter should be considered especially when there are atrioventricular conduction disturbances, prior to or concomitant with ventricular dysfunction or skeletal myopathy.30 Other diagnostic criteria are used for familial forms (Table S4, Supplement 8).24
Genetic diagnosisGenetic testing is recommended (class I) for patients diagnosed with DCM and significant cardiac conduction disease ((i.e., first-, second-, or third-degree heart block) and/or a family history of premature unexpected SD,2 and in patients with a familial form of DCM or in sporadic DCM with clinical clues suggestive of a particular/rare genetic disease.24 Genetic testing should be considered in the most clearly affected person in a family (level of evidence A)23 (Table 2).
Supplement 9 lists some manifestations of rare genetic forms of DCM that should prompt molecular diagnosis.
Genetic testing has a positive result in 30-40% of cases of DCM,31 higher in familial than in isolated cases (25-40% vs. 10-25%).23
Arrhythmogenic right ventricular cardiomyopathyClinical diagnosisArrhythmogenic right ventricular cardiomyopathy (ARVC) is defined histologically by the progressive replacement of ventricular myocardium with adipose and fibrous tissue often confined to a ‘triangle of dysplasia’ comprising the right ventricular inflow, outflow, and apex.22 It is defined by the presence of right ventricular dysfunction (global or regional), with or without left ventricular disease, in the presence of histological evidence for the disease and/or abnormalities on ECG, echocardiography or cardiac magnetic resonance (CMR) (Table S5, Supplement 10).32
Although structural abnormalities predominate in the right ventricle, it is now known that involvement can be biventricular or even mainly in the left ventricle.33 There should be a high index of suspicion for this condition when non-ischemic late enhancement sparing the subendocardium is seen on CMR together with T-wave abnormalities on the ECG and ventricular arrhythmias, particularly in the presence of a family history of SD.34
Diagnosis in familial casesWhen ARVC has been diagnosed in an index case, diagnosis in first-degree relatives requires only one of the following criteria: T-wave inversion in V1, V2 and V3 (over the age of 14 years); late potentials; VT with left bundle branch block morphology or >200 premature ventricular contractions in 24 hours; or mild right ventricular global or segmental dilatation or dysfunction.32
Genetic diagnosisComprehensive or targeted genetic testing can be useful for patients satisfying the diagnostic criteria for ARVC (class IIa2,3/level A23), and may be considered in possible cases (class IIb).2 The positivity rate for genetic testing in ARVC is usually around 50%35 (Table 2).
Restrictive cardiomyopathyClinical diagnosisRestrictive cardiomyopathy (RCM) is rare and can be idiopathic, familial or secondary to systemic disorders. Characterized by a restrictive physiology, it is usually detected by echocardiography, which shows normal or reduced ventricular volumes and wall thicknesses that are not significantly increased (although wall thickness may be increased in infiltrative disease).22
Genetic diagnosisGenetic testing may be considered for patients with HCM based on clinical history, family history, and electrocardiographic/echocardiographic findings (class IIb2/level B23) (Table 2).
Before genetic study is performed, differential diagnoses and specific diagnostic tests should be considered as listed in Supplement 11.
Left ventricular non-compactionClinical diagnosisLeft ventricular non-compaction (LVNC) is characterized by prominent left ventricular trabeculae and deep intertrabecular recesses with blood flow from the ventricular cavity and no communication with the coronary tree. Two layers of myocardium can be distinguished, compacted and non-compacted.22 In some individuals, LVNC is associated with ventricular dilatation and systolic dysfunction.22
It is not clear whether LVNC is a separate primary cardiomyopathy or merely a phenotypic trait shared by other cardiomyopathies and other pathological (neuromuscular and mitochondrial diseases and myopathies) and physiological conditions, including pregnancy and sports participation.23,36,37
There are various diagnostic criteria based on imaging studies (Table S6, Supplement 12).38–44
A diagnosis of cardiomyopathy is more likely when the quantitative short-axis echocardiographic criteria of Jenni38 or Jacquier43 are fulfilled together with one of the following factors: another affected family member (or family history of cardiomyopathy); ventricular dysfunction or wall motion abnormalities; symptoms or complications; neuromuscular disease; or one of several potentially causative genetic variants found in families with LVNC.45,46
Genetic diagnosisGenetic testing may be considered for patients with a clinical diagnosis of LVNC based on clinical history, family history, and clinical (especially neuromuscular disease), electrocardiographic and echocardiographic findings (class IIa).2 It is not recommended in individuals with isolated LVNC or those without other abnormalities in ventricular structure or function, symptoms, or family history (Table 2).
Family screening in cardiomyopathiesDetails of clinical and molecular assessment of relatives of patients with cardiomyopathies are given in Table S7, Supplement 13.
Hereditary aortopathiesHereditary diseases of the aorta are a heterogeneous group of disorders characterized by aneurysms and/or dissection in one or more aortic segments, most often between the aortic annulus and the diaphragm. Depending on the presence or otherwise of manifestations in other organs, hereditary aortopathies can be classified as syndromic or non-syndromic. Associated genetic variants mainly affect extracellular matrix homeostasis, transforming growth factor-beta (TGF-β) signaling, and vascular smooth muscle cell contractility.47,48
In patients with thoracic aorta aneurysms or dissection, screening of first-degree relatives is recommended to identify possible familial forms (class I). Once a familial form is strongly suspected, it is recommended to refer the patient to a geneticist for family investigation and molecular testing (class I). In non-syndromic familial cases, screening for aneurysms should be considered throughout the arterial tree, including cerebral arteries (class IIa).49
The main syndromes associated with aortic aneurysms or dissection and indications for imaging of the aorta are listed in Tables S8.1, S8.2 and S8.3, Supplement 14.
Familial hypercholesterolemiaAlthough knowledge of familial hypercholesterolemia (FH) has improved in recent decades, this common genetic disease, which though potentially fatal is treatable, remains underdiagnosed and undertreated.50 Early diagnosis is essential to begin appropriate individualized treatment in order to prevent premature atherosclerotic disease, as well as to identify affected relatives and thus reduce the burden of cardiovascular disease in these families.
Clinical diagnosisThere are two main sets of diagnostic criteria for FH: those of the Simon Broome Register Group51 and those of the Dutch Lipid Clinic Network.52 The criteria most often used in Portugal are adapted from those of the Simon Broome Register, as follows:
Probable familial hypercholesterolemia- (a)
Children under 16 years of age: >260 mg/dl or low-density lipoprotein (LDL) cholesterol >155 mg/dl; adults: total cholesterol >290 mg/dl or LDL cholesterol >190 mg/dl and
- (b)
family history of myocardial infarction before 50 years of age in grandparents or uncles/aunts or before 60 years of age in parents, siblings or children and/or
- (c)
family history of raised total cholesterol (>290 mg/dl in adults and >260 mg/dl in children under 16 years of age) in parents, siblings or children; or total cholesterol >290 mg/dl in grandparents or uncles/aunts.
Cholesterol levels should be measured on two different occasions, preferably 3-6 months after implementation of appropriate lifestyle changes.
Confirmed familial hypercholesterolemia- (a)
The above criteria plus tendon xanthomas in first-degree (parents, siblings or children) or second-degree (grandparents or uncles/aunts) relatives or
- (b)
presence of a pathogenic or likely pathogenic variant in one of three genes associated with HF: LDLR, APOB, or PCSK9.
Genetic testing should be performed in individuals with definite or probable FH, as well as for their at-risk relatives, as recommended by the Journal of the American College of Cardiology expert panel.53
The test should include the genes LDLR, APOB and PCSK9, and whenever possible the genes associated with phenocopies of FH: LDLRAP1, APOE, LIPA, ABCG5 and ABCG8.53
Genetic testing provides prognostic information that can lead to more accurate risk stratification. Cascade screening of at-risk relatives is highly effective in identifying affected individuals who require appropriate treatment.
Individuals with confirmed FH, particularly those with homozygous FH, should be referred to a specialist in the condition. All FH patients should be offered a regular structured review that is carried out at least annually, since this can reduce cardiovascular morbidity and mortality through lifestyle interventions and prompt appropriate therapeutic measures.54
Congenital heart diseaseCongenital heart disease (CHD) is the umbrella term for cardiovascular malformations present from birth, which are found in 1-1.2% of all live births.55 The actual prevalence is difficult to determine, since not all cases are diagnosed early, but it has been estimated at 13.1/1000 children and 6.1/1000 adults.56 Around 90% of cases are sporadic. Most children with CHD now survive to adulthood, although significant proportions require one or more surgical interventions or develop complications such as arrhythmias or heart failure.
The main etiological factors in CHD and methods of genetic diagnosis are listed in Supplement 15.
An increasing number of genetic variants associated with CHD have been identified in the last decade, including sporadic, hereditary, syndromic and non-syndromic forms. A definite genetic diagnosis may, for example, identify a non-cardiac phenotype, which will require specific management and treatment and will improve genetic counseling. Supplement 15 includes some common examples of syndromic CHD (Table S9.1) and data on the risk of recurrence in some CHD (Tables S9.2 and S9.3).
Pulmonary arterial hypertensionClinical diagnosisPulmonary arterial hypertension (PAH) is defined as mean pulmonary arterial pressure >20 mmHg associated with pulmonary artery wedge pressure ≤15 mmHg and pulmonary vascular resistance ≥3 Wood units at rest, measured by right heart catheterization.57
Around 70-80% of patients with PAH inherited in an autosomal dominant pattern58 and 10-20% with sporadic or idiopathic PAH59 have variants in the BMPR2 gene, which codes for a protein in the TGF-β family. Penetrance is greater in females in these variants.60
Other genes have been associated with PAH, including TBX4, ATP13A3, GDF2, SOX17, AQP1, ACVRL1, SMAD9, ENG, KCNK3 and CAV1.58 Variants in EIF2AK4 are associated with pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis.61
Genetic diagnosisGenetic testing and counseling should be offered to patients with hereditary or sporadic/idiopathic PAH. Genetic testing can identify asymptomatic carriers, but since penetrance is incomplete, it is currently impossible to predict who will develop the disease. Echocardiographic monitoring should be considered for such individuals.59,62 Identification of genetic variants in EIF2AK4 can avoid the need for pulmonary biopsy.58
Pre-implantation genetic diagnosis of PAH can enable selection of embryos for medically assisted reproduction.63
Sudden cardiac deathDefinitionsSD is defined as a non-traumatic, unexpected fatal event occurring within one hour of the onset of symptoms in an apparently healthy subject. If death is not witnessed, it is defined as death when the victim was in good health 24 hours before the event.9
Sudden arrhythmic death syndrome (SADS) is defined as SD when the cause of death is unknown or uncertain after autopsy, which is the case in 25-50% of young SD.64,65
SD in children aged under one year is known as sudden infant death syndrome. In this age-group, there is a complex relationship between various factors that can trigger SD, including a critical development period of the autonomic nervous system, exogenous stressors such as sleeping position, and the inherent vulnerability of the infant, including genetic factors.66
SD accounts for 15-25% of mortality in the general population and its incidence increases significantly with age.67–70 Over the age of 40 years, most cases are due to coronary artery disease,71 while hereditary cardiac conditions such as cardiomyopathies and channelopathies are more common in younger individuals.69
Primary electrical diseases such as LQTS, BrS and CPVT are not easy to identify postmortem, and postmortem genetic testing (molecular autopsy) is particularly useful in these cases. In SADS, genetic testing can identify the cause of death in an additional 20-30% of cases. It should be noted that genetic variants associated with channelopathies, especially LQTS and CPVT, have been identified in around 20% of patients previously diagnosed with epilepsy.72
Autopsy and postmortem genetic testingIn cases of SD, it is important to diagnose hereditary heart disease so that at-risk living relatives (asymptomatic carriers) can be identified, in order to intervene early and to modify the course of their lives.70
An autopsy is recommended to investigate the causes of SD (class I).9 The guidelines for autopsies in these cases were recently updated by the Association for European Cardiovascular Pathology.73 Ideally, information should be collected on the personal and family medical history of the deceased and the circumstances of death. The cardiac examination should be performed by an experienced pathologist and biological material should be obtained for genetic study, if performed. Handling of this material requires the consent of the deceased’s family.74
The results of the autopsy should be communicated to the family. Referral networks should be in place for appropriate clinical assessment of relatives. Postmortem genetic testing should only be performed after relatives receive genetic counseling75 and is indicated when hereditary heart disease is suspected, whether suggested by autopsy results or in cases of SADS (class IIa).9
In the context of SADS, genetic study should include genes associated with channelopathies and cardiomyopathies. Although the latter cause structural changes in the myocardium, these may be subtle and undetected on autopsy.76
As by definition there is no identified phenotype in cases of SD, even if a genetic variant is identified its pathogenicity often cannot be established. It is thus important to integrate this information with the results of familial screening.72
Screening of relativesAccording to the European Society of Cardiology guidelines, whether or not molecular autopsy is performed, screening of first-degree relatives of SD victims is recommended (class I).9 Screening should initially include clinical assessment, ECG, stress testing and echocardiography and then, depending on the level of clinical suspicion, CMR, 24-h Holter, and provocative testing with ajmaline/flecainide.9
In the absence of a definitive diagnosis, following a thorough assessment (which may include genetic study), first-degree relatives should be monitored periodically until adulthood, by which time most relevant diseases will have reached phenotypic expression.9 When the results of autopsy and molecular study are negative, the event rate in relatives of SD victims appears to be low.77
Conflicts of interestThe authors have no conflicts of interest to declare.
The following are Supplementary data to this article:
Please cite this article as: Sousa A, Moldovan O, Lebreiro A, Bourbon M, António N, Rato Q, et al. Recomendações para a realização de testes genéticos em cardiologia – revisão das principais diretrizes internacionais. Rev Port Cardiol. 2020;39:597–610.
