
A síndrome do QT longo congénito (SQTLC) é uma doença hereditária rara, com uma incidência de uma em cada 2000 pessoas, caracterizada por uma repolarização ventricular prolongada e por taquiarritmias ventriculares malignas.
Reportamos o caso de uma doente de 30 anos, com diagnóstico prévio de síncope neurocardiogénica, em que identificamos a SQTLC. A doente implantou cardioversor‐desfibrilhador implantável por taquicardia ventricular polimórfica apesar da terapêutica com bloqueador beta.
Foram identificadas nesta doente três mutações em heterozigotia nos genes KCNH2, KCNQ1 e SCN5A, achado raro que lhe confere um pior prognóstico.
Congenital long QT syndrome (LQTS) is a rare hereditary disease, with an incidence of 1 in 2000, characterized by prolonged ventricular repolarization and malignant ventricular tachyarrhythmias. We report the case of a 30‐year‐old woman, previously diagnosed with neurocardiogenic syncope, in whom LQTS was identified. The patient received an implantable cardioverter‐defibrillator due to polymorphic ventricular tachycardia under beta‐blocker therapy. Molecular genetic testing identified three mutations in heterozygosity in the KCNH2, KCNQ1 and SCN5A genes, which is a rare finding and is associated with worse prognosis.
A síndrome do QT longo congénito (SQTLC) é uma doença hereditária rara, com uma incidência de uma em cada 2000 pessoas, caraterizada por uma repolarização ventricular prolongada e por taquiarritmias ventriculares malignas, carateristicamente taquicardia ventricular (TV) polimórfica ou torsades de pointes, que podem culminar em síncope ou morte súbita1–3. Os eventos arrítmicos surgem habitualmente por volta dos 12 anos.
Caso clínicoDoente do género feminino, de 30 anos de idade, avaliada em consulta de cardiologia pediátrica por síncopes de repetição. Apresentava síncopes de repetição desde a infância, tendo sido primeiramente diagnosticado epilepsia. Realizou teste de tilt com registo de resposta vasodepressora; iniciou midodrina, sem melhoria clínica. Desconhecem‐se registos eletrocardiográficos dessa altura.
Foi orientada do centro de saúde para consulta de cardiologia neste centro por manter síncope recorrente. A doente referia episódios de pré‐síncope e síncope, ocasionalmente precedidos de palpitações e por vezes precipitados pelo ortostatismo.
Não apresentava outros antecedentes médicos relevantes. Na história familiar referia apenas morte súbita da mãe com 46 anos de idade, com antecedentes médicos de diabetes mellitus tipo 2, tendo o exame anatomopatológico demonstrado lesões de cardiopatia isquémica crónica.
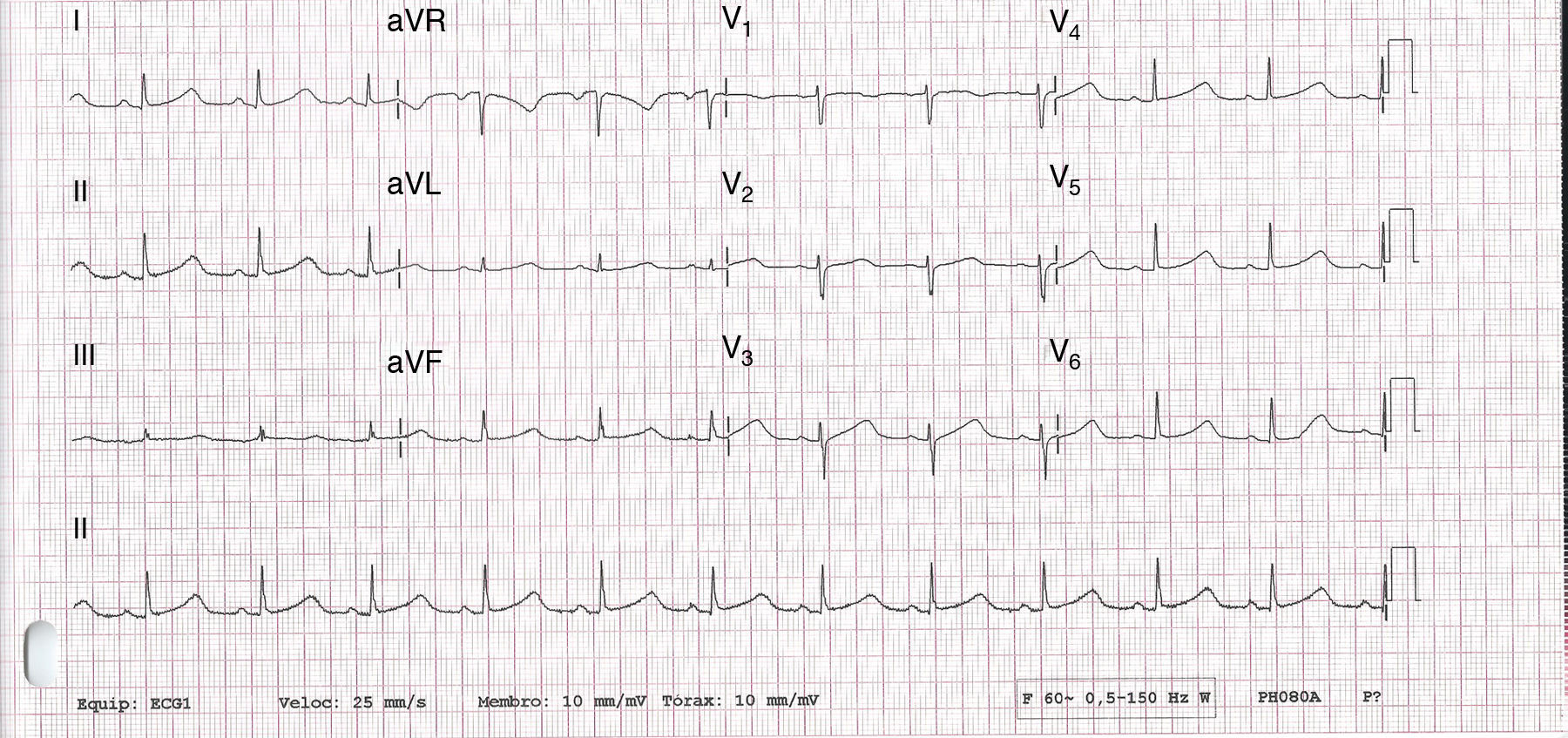
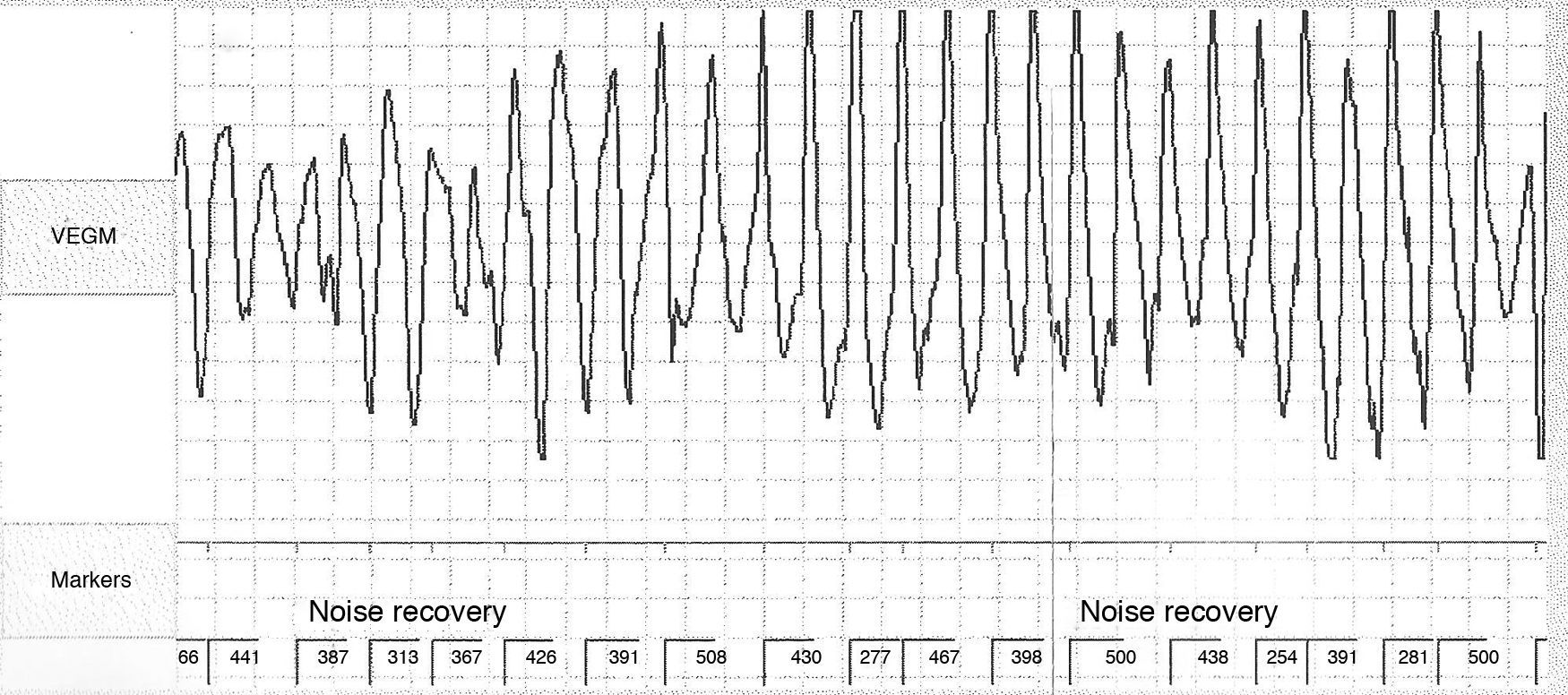
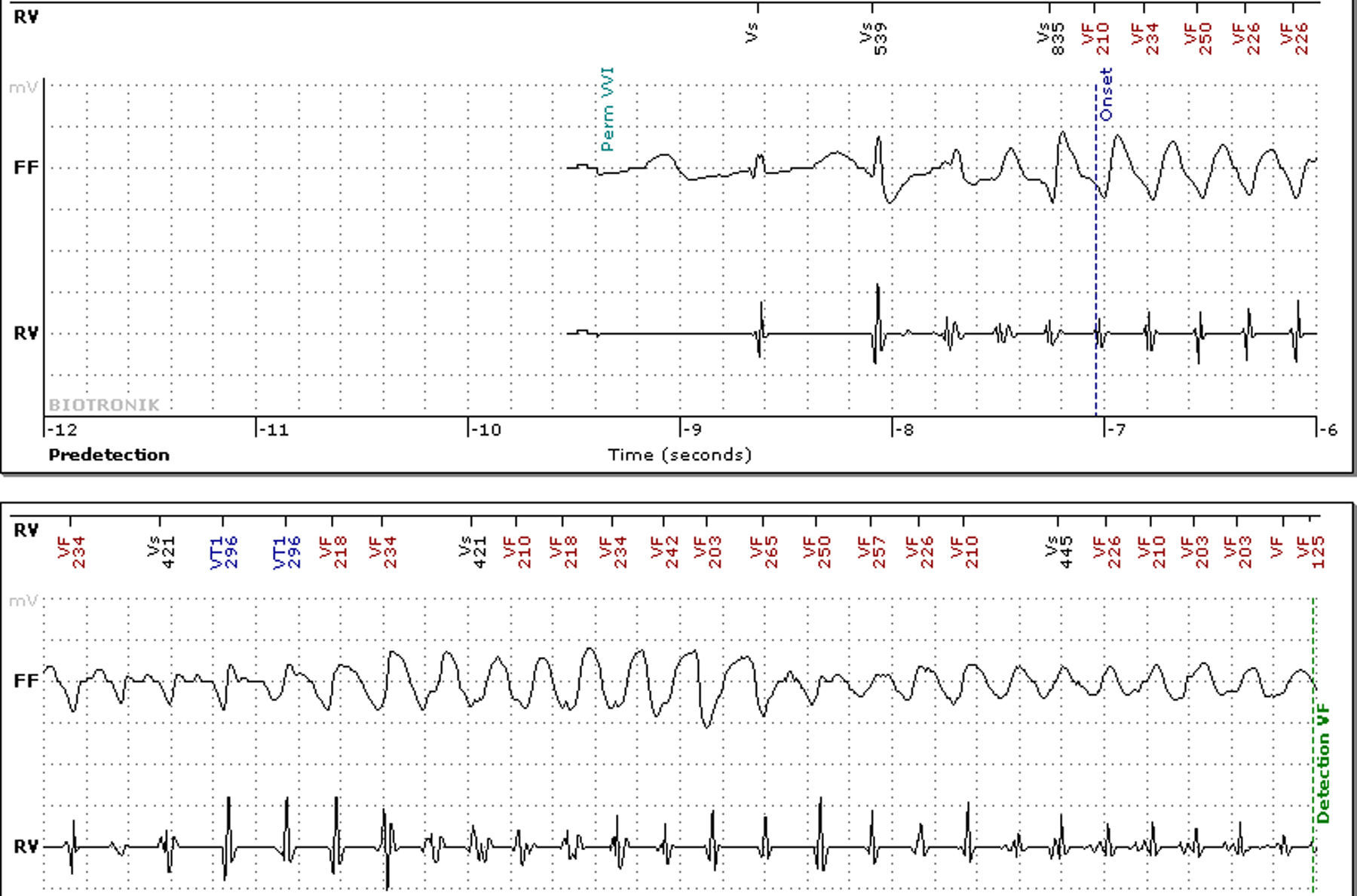
Não apresentava alterações analíticas e o ecocardiograma transtorácico não evidenciou alterações de relevo. O eletrocardiograma de 12 derivações (ECG) realizado nas consultas de cardiologia registava ritmo sinusal e prolongamento significativo do intervalo QTc (variando entre 500 e 565ms) (Figura 1). Encontrava‐se medicada com carvedilol 12,5mg bid, não se conseguindo titular a dose por hipotensão sintomática. Atendendo ao resultado do teste de tilt anteriormente realizado e sintomas dúbios, foi decidido implantar‐se registador de eventos. A doente relatou vários episódios de pré‐síncope que corresponderam a períodos de taquicardia sinusal e num dos episódios foi registada TV polimórfica não sustida (Figura 2). Foi decidido implantar‐se cardioversor‐desfibrilhador implantável (CDI) e cerca de um mês após o procedimento registou‐se na monitorização remota choque apropriado por TV polimórfica (Figura 3). Alterada terapêutica bloqueadora beta com início de propanolol.
.")


No teste genético molecular foi encontrada em heterozigotia a mutação c.785delG no exão 4 do gene KCNH2. Esta mutação leva à formação de um codão STOP prematuro na posição 359 da proteína (p.Gly262AlafsX98). Foram ainda encontradas mais duas alterações:
- ‐
c.535G>A, em heterozigotia, no exão 3 do gene KCNQ1. Esta alteração leva à substituição de um aminoácido na posição 179 da proteína (p.Gly179Ser).
- ‐
c.3068G>A, em heterozigotia, no exão 17 do gene SCN5A. Esta alteração leva à substituição de um aminoácido na posição 1023 da proteína (p.Arg1023His).
A síndrome do QT longo é uma doença genética caraterizada por um prolongamento significativo do intervalo QT, predispondo a arritmias ventriculares, manifestando‐se clinicamente com síncope, convulsões ou morte súbita em doentes com coração estruturalmente normal1,2. Face à ausência de cardiopatia estrutural e elevada incidência de síncopes de etiologia vasovagal e epilepsia nos jovens, o diagnóstico é frequentemente tardio e erróneo4.
De acordo com as recomendações de um consenso de peritos o diagnóstico de síndrome de QT longo baseia‐se nos seguintes critérios: presença de um score de risco de SQTL ≥3,5 na ausência de outras causas secundárias de prolongamento do intervalo QT e/ou presença de uma mutação patogénica num dos genes do SQTL ou um intervalo QTc ≥500ms em ECG repetidos. O diagnóstico poderá ser realizado na presença de um intervalo QTc entre 480‐499ms em ECG repetidos em doentes com síncope inexplicada na ausência de causas secundárias e na ausência de mutações patogénicas1.
O score de Schwartz estima a probabilidade de diagnóstico de SQTL com base em critérios clínicos, eletrocardiográficos e história familiar. Apresenta essencialmente duas limitações: apesar da sua alta sensibilidade é pouco específico e exclui o teste genético como critério de diagnóstico, não identificando 30% dos portadores assintomáticos5–8. Já foram identificadas mutações em vários genes e são reportados frequentemente novos casos, sendo que as mais frequentes são mutações nos genes KCNQ1 (SQTLC Tipo 1), KCNH2 (SQTLC Tipo 2) e o SCN5A (SQTLC Tipo3)1–3,7,9. As duas primeiras mutações conduzem a perda de função dos canais de potássio e a última a ganho de função dos canais de sódio, ocorrendo alteração nas correntes iónicas com subsequente instabilidade da membrana. A maior parte dos casos apresenta um padrão de hereditariedade autossómica dominante.
Através das caraterísticas clínicas, nomeadamente os triggers de taquiarritmias ventriculares, e do padrão eletrocardiográfico do prolongamento do intervalo QT é possível inferir corretamente acerca do genótipo em 70‐90% dos casos10. A SQTLC1 caracteriza‐se por uma onda T ampla e larga e, devido à sua dependência adrenérgica, os eventos arrítmicos surgem durante o exercício ou emoções. Na SQTLC2 a onda T é pouco ampla e entalhada e os triggers mais frequentes são emoções e estímulos auditivos. A SQTLC3 caracteriza‐se por um prolongamento do intervalo QT essencialmente através de um prolongamento do segmento ST isoelétrico e os eventos surgem habitualmente em repouso ou durante o sono, denunciando a sua pouca sensibilidade ao sistema simpático e a sua questionável resposta aos bloqueadores beta7,10.
Quando é registado um prolongamento do intervalo QT é importante excluir iatrogenia farmacológica, hipotiroidismo e distúrbios eletrolíticos. Apesar do QT longo poder surgir no contexto de iatrogenia farmacológica, esta pode também evidenciar uma SQTLC até então oculta.
O teste genético, que identifica a mutação em 50‐75% dos casos, assume um papel importante na confirmação do diagnóstico, na identificação dos portadores assintomáticos, na estratificação de risco de eventos arrítmicos e no aconselhamento genético7. A identificação dos silent carriers é fundamental pois são doentes que têm um risco elevado de arritmias ventriculares graves.
Os doentes de maior risco são os doentes com SQTL1 e SQTL2 com QTc>500ms e doentes do género masculino com SQTL3, independentemente do intervalo QTc. A presença de mutações digénicas e mutações compostas são encontradas em 8% dos doentes com SQTL e associam‐se a um fenótipo mais grave12,13.
A nossa doente apresenta três mutações identificadas em três genes distintos. A mutação p.Gly262AlafsX98 no gene KCNH2 foi previamente descrita em doentes com síndrome QT longo (Human Gene Mutation Database [HGMD] – CD070027) e está estabelecida como causa desta patologia14. As mutações p.Gly179Ser no gene KCNQ1 e p.Arg1023His no gene SCN5A também já foram descritas em doentes com síndrome QT longo (HGMD – CM002321 e CM054857, respetivamente)14.
Em todos os doentes com diagnóstico clínico ou molecular estão contraindicados fármacos que prolonguem o intervalo QT e devem ser identificados e tratados eventuais distúrbios hidroeletrolíticos. Os bloqueadores beta estão recomendados em doentes assintomáticos com intervalo QTc ≥470ms e/ou doentes sintomáticos com síncope ou arritmias ventriculares (classe I), podendo ser equacionados em doentes assintomáticos com QTc<470ms (classe IIa). A desnervação simpática deve ser considerada nos doentes de alto risco não candidatos a CDI e nos doentes com arritmias ventriculares apesar de medicados com bloqueadores beta ou com intolerância aos mesmos (classe I)1. Em todos os doentes reanimados de paragem cardiorrespiratória e em doentes com TV ou síncope sob bloqueador beta está indicada a implantação de CDI1,2. Nos doentes com SQTL3 e intervalo QTC>500ms os bloqueadores dos canais de sódio podem ser eficazes, associados aos bloqueadores beta1. No que diz respeito ao bloqueador beta mais eficaz nestes doentes, existe alguma evidência de que o nadolol e o propanolol possam ser mais seguros e eficazes11.
ConclusõesA síndrome do QT longo é uma doença hereditária rara em doentes sem cardiopatia estrutural, que se associa a um risco elevado de taquiarritmias ventriculares malignas e morte súbita. O CDI é a estratégica terapêutica indicada após ressuscitação de morte súbita ou persistência de sintomas apesar de terapêutica com bloqueador beta. Este caso descreve uma doente com síndrome do QT longo com identificação genética de três mutações em três genes distintos, uma situação rara, que condiciona um mau prognóstico.
Responsabilidades éticasProteção de pessoas e animaisOs autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dadosOs autores declaram que não aparecem dados de pacientes neste artigo.
Direito à privacidade e consentimento escritoOs autores declaram que não aparecem dados de pacientes neste artigo.
Conflito de interessesOs autores declaram não haver conflito de interesses.
