
In up to one-third of cases of sudden death, the medico-legal autopsy finding is inconclusive, and the option to perform a molecular autopsy is covered in international guidelines. The importance of postmortem genetic testing lies in its ability to identify hereditary diseases, often those with an autosomal dominant transmission pattern, and, through consultations and screening of relatives, to identify family members with a pathogenic mutation, who are often asymptomatic, providing an opportunity to change the course of their lives. The authors present three clinical cases that highlight the importance of postmortem genetic studies and family studies, as well as the integration of the data obtained in a cardiology consultation, which may be for arrhythmology, coronary disease or cardiomyopathy, depending on the specific condition. This could modify the course of the disease in many relatives.
Em até um terço dos casos de morte súbita, a autópsia médico-legal é “branca”, estando já contemplada nas recomendações internacionais a possibilidade da realização de autópsia molecular. A importância do teste genético post mortem prende-se com a identificação de doenças hereditárias, frequentemente doenças com padrão de transmissão autossómico dominante, sendo possível, através de consulta e rastreio de parentes, identificar elementos na família com a doença, não raramente portadores assintomáticos de uma mutação, havendo espaço para alterar o curso de vida dos mesmos. Os autores apresentam três casos clínicos que reforçam o quão importante é o estudo genético post mortem, assim como o estudo familiar e a integração dos dados numa consulta de cardiologia, seja arritmologia, consulta de doença coronária ou miocardiopatias, dependendo da patologia específica, podendo modificar o curso da doença em muitos parentes.
Cardiovascular mortality has decreased in the last 20 years, as a result of preventive measures to reduce the burden of coronary artery disease and heart failure.1 However, cardiovascular disease is still responsible for 17 million deaths every year worldwide, 24% of which are sudden cardiac death (a non-traumatic, unexpected fatal event occurring within one hour of the onset of symptoms in an apparently healthy subject, or if not witnessed, when the victim was in good health 24 hours before the event). Sudden arrhythmic death syndrome is defined as sudden death with inconclusive autopsy and toxicology and histology investigations, a structurally normal heart and exclusion of non-cardiac etiologies.1 In up to one-third of cases of sudden death, the medico-legal autopsy finding is inconclusive, and the option to perform a molecular autopsy is covered in international guidelines.1 The importance of postmortem genetic testing lies in its ability to identify hereditary diseases, often those with an autosomal dominant transmission pattern, and, through consultation and screening of relatives, to identify family members with a pathogenic mutation, who are often asymptomatic, providing an opportunity to change the course of their lives. Genetic study identifies channelopathies in 35-50% of cases of sudden arrhythmic death, and pathogenic mutations can be identified in 25-35% of their relatives.1–4
We present three clinical cases that highlight the importance of postmortem genetic studies and family studies, as well as the integration of the data obtained in a cardiology consultation, which may be for arrhythmology, coronary disease or cardiomyopathy, depending on the specific condition. This could modify the course of the disease in many relatives.
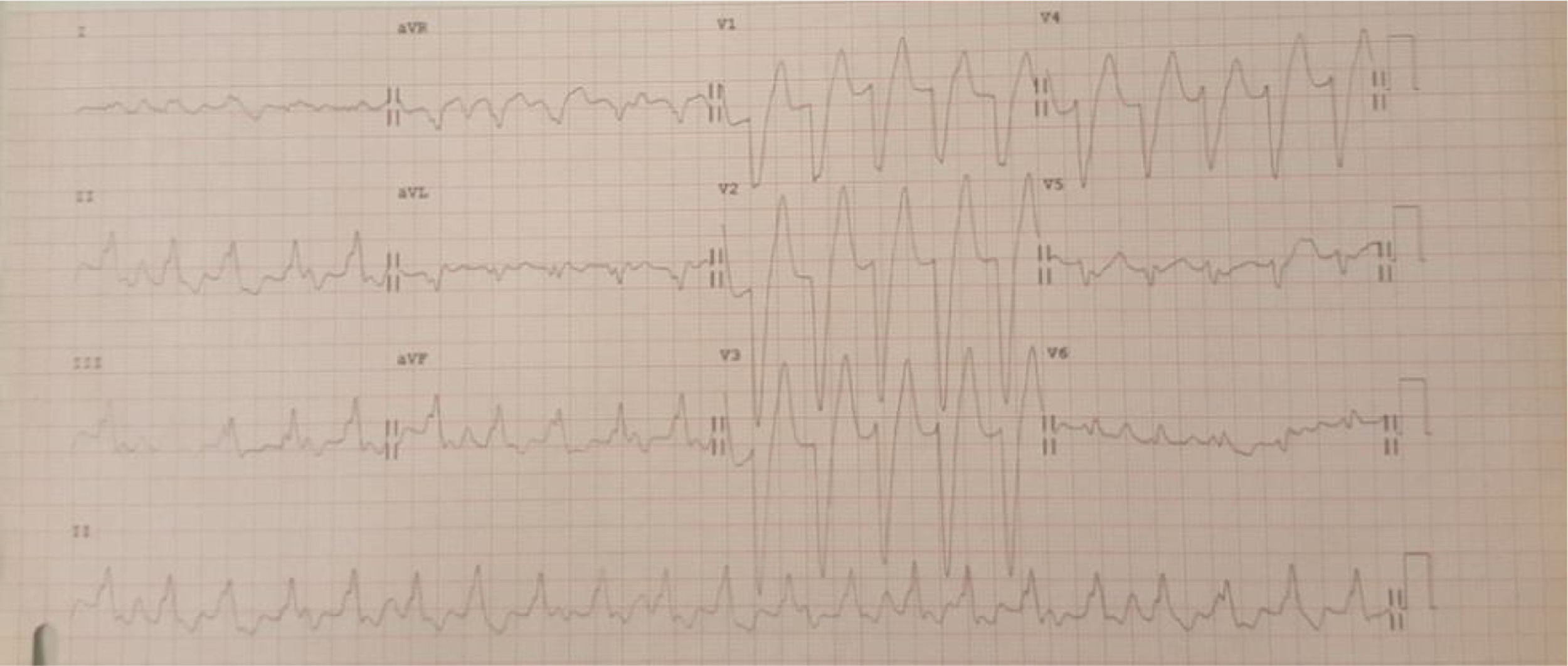
Case 1 (2010)The first case is of a 15-year-old female with episodes of palpitations without syncope. Episodes of slow monomorphic ventricular tachycardia were documented (Figure 1). Transthoracic echocardiography revealed normal sized cardiac chambers and preserved biventricular systolic function, but with left ventricular hypertrabeculation. Cardiac magnetic resonance imaging was then performed which diagnosed left ventricular noncompaction. Electrophysiological study documented branch-to-branch reentry ventricular tachycardia; it was decided not to perform ablation because of intraventricular conduction delay on the baseline electrocardiogram (ECG) and the possible need for pacing. The patient received an implantable cardioverter-defibrillator (ICD), but in 2014 she was admitted to the emergency department following cardiac arrest in ventricular fibrillation after several unsuccessful ICD shocks.
Case 2 (2014)
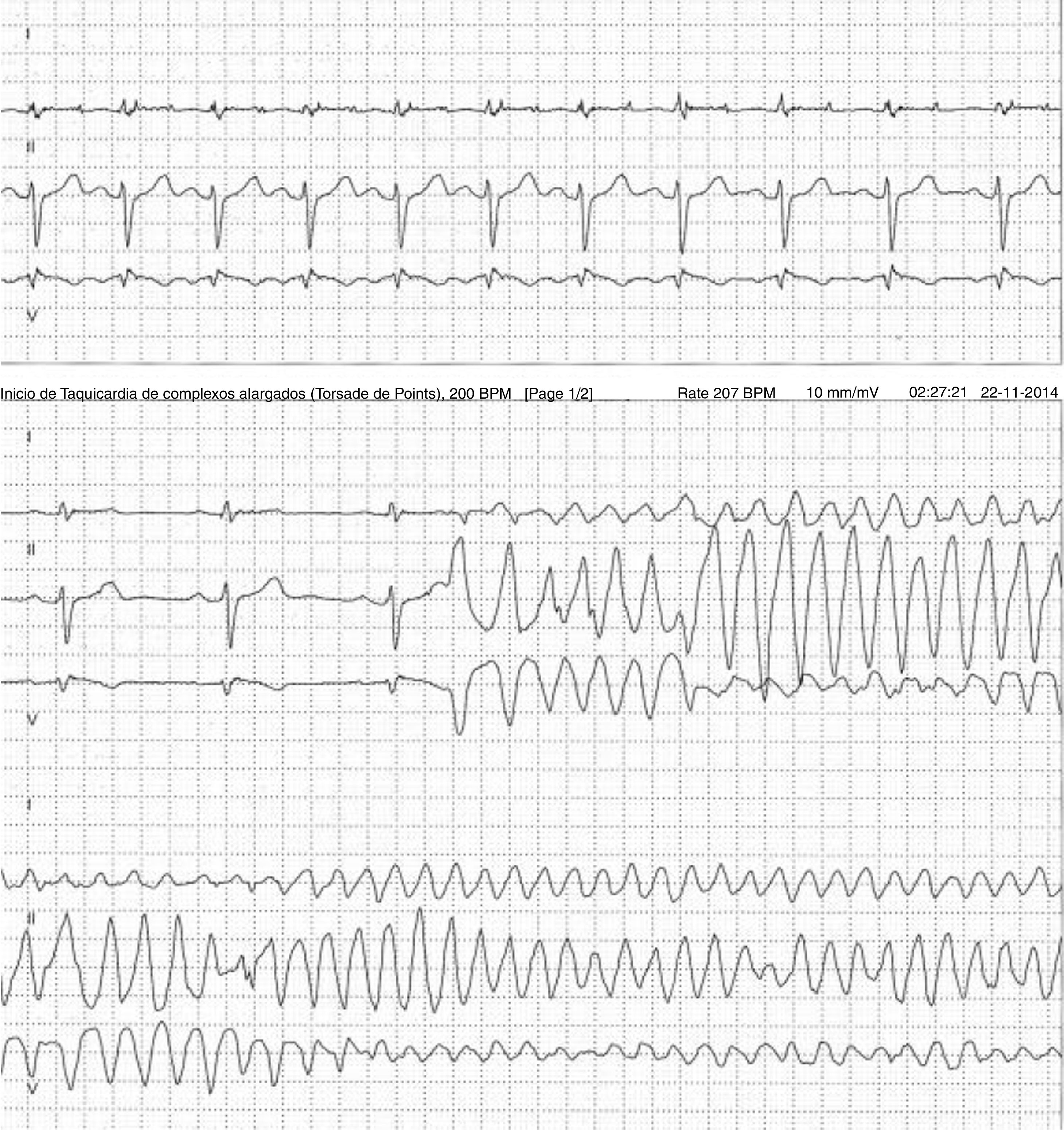
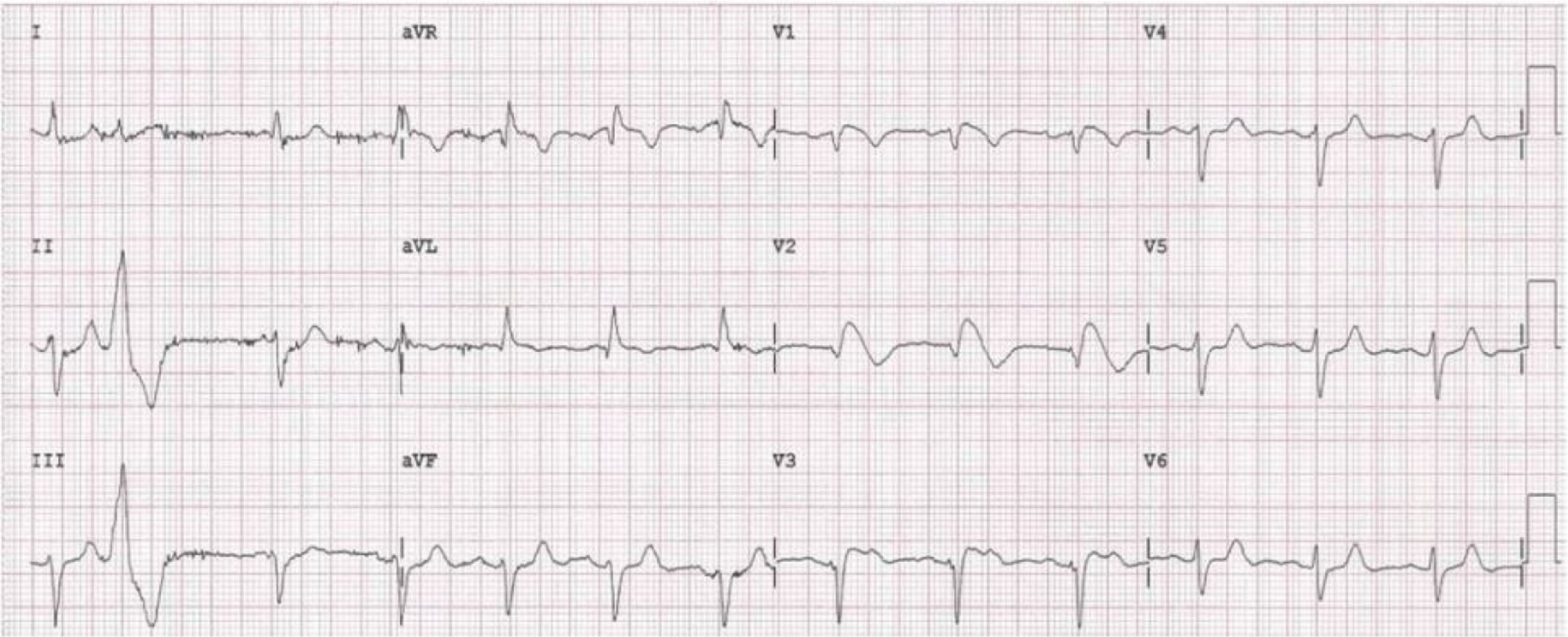
A 59-year-old female patient, who knew of no relevant family history and had episodes of palpitations without syncope, underwent transthoracic echocardiography, which was normal, and 24-hour Holter monitoring at the request of her general practitioner. The latter revealed polymorphic ventricular tachycardia triggered by an R-on-T ventricular extrasystole (Figure 2) and followed by asystole and atrioventricular block, lasting for around 4 min. When the Holter monitoring was reviewed at the doctor's office, the patient was sent to the emergency department, where laboratory tests were normal and the ECG showed type 1 Brugada pattern (Figure 3). An ICD was placed and she underwent genetic testing, which identified the 4720G>A variant in the SCN5A gene (MIM 600163), classified as pathogenic, in heterozygosity. An arrhythmology consultation was scheduled for her relatives, of whom only her two sons were assessed; according to the patient her relationship with the rest of her family was poor and they would not be interested in attending a consultation.


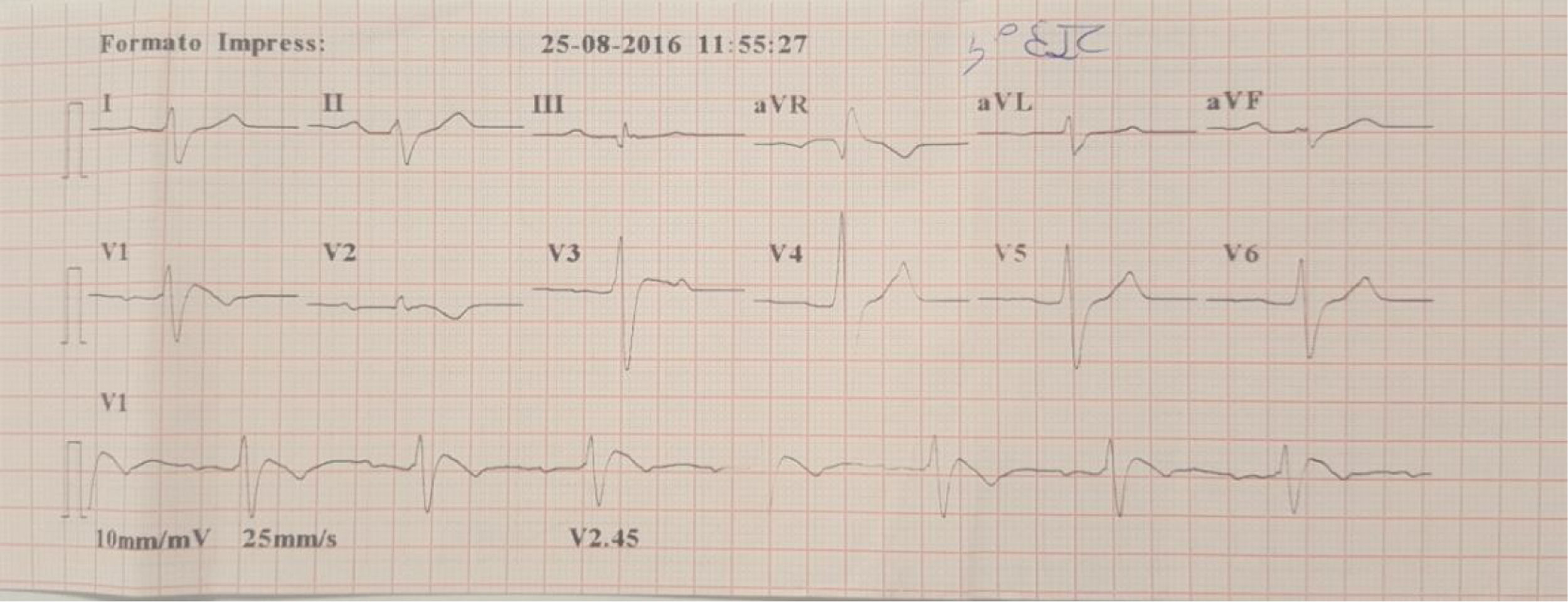
A family were referred for clinical assessment in cardiology consultations. They were relatives of a 30-year-old man with no known relevant family history who was asymptomatic until August 2015, when he suffered sudden death while driving. The medico-legal autopsy found no evidence of cause of death. The medical examiner's office requested genetic testing as part of a molecular autopsy, which identified the 4720G>A variant of the SCN5A gene in heterozygosity. When the decedent's relatives were informed of this result, they were referred for arrhythmology consultations and his parents and three sisters were assessed. The parents are asymptomatic with no alterations on baseline ECG. After genetic counseling, genetic testing was performed which identified the mutation in the mother. One of the sisters reported episodes of syncope with seizures; her ECG, with V1 and V2 in the second, third and fourth intercostal spaces, showed sinus rhythm with no alterations, but pharmacological provocation with flecainide produced type 1 Brugada pattern (Figure 4). After discussion with the patient, it was decided to place an ICD. Genetic testing identified the same mutation as in her mother and deceased brother. Another sister is asymptomatic, with normal baseline ECG, but carrying the same mutation, while the third sister is also asymptomatic and with normal baseline ECG, and does not have the mutation.

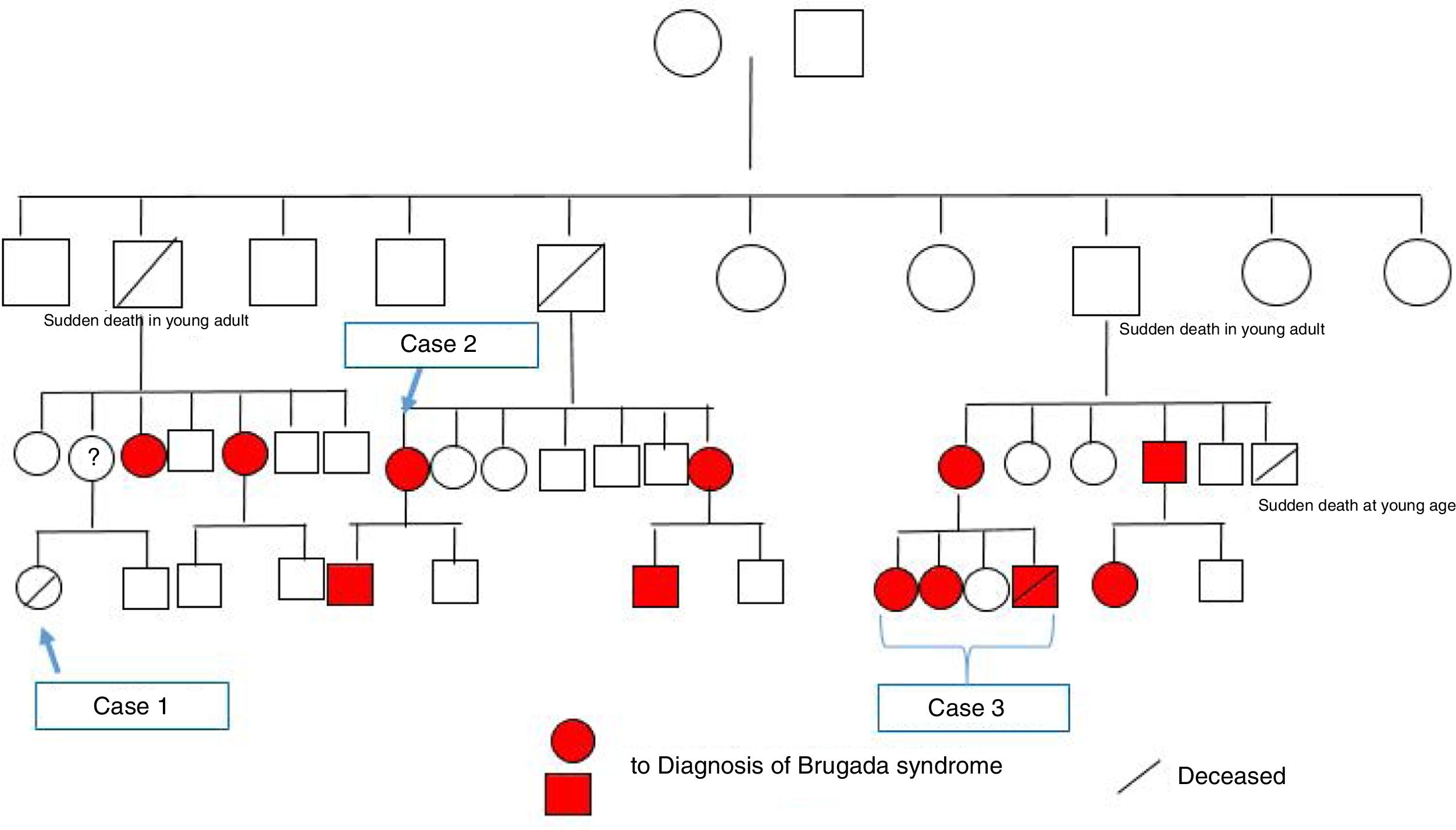
Since both the surname and the mutation identified in this case were the same as in case 2, and since the latter family were now on speaking terms with their relatives, we came to the conclusion that both cases were members of the same family. They also reported that a distant female relative had suffered sudden death at a young age. After construction of the family tree (Figure 5), it was discovered that this relative was the girl described in case 1 above.

Through arrhythmology and medical genetics consultations, an additional 11 family members with the mutation were identified, six of whom have been found to have a spontaneous type 1 Brugada ECG, and further studies are ongoing.
DiscussionThe incidence of sudden cardiac death increases significantly with age, like that of coronary artery disease. Sudden cardiac death is rare before the age of 35 years, when the most frequent causes are myocarditis, cardiomyopathy, premature coronary artery disease, congenital coronary artery anomalies and channelopathies.1,5 According to a recent study on sudden death in athletes, sudden arrhythmic death syndrome was the leading cause, with a structurally normal heart reported in 56% of those aged under 18 years, 44% of those aged between 18 and 35 and 26% of those aged >35 years.6
Brugada syndrome is diagnosed on the basis of a specific electrocardiographic pattern: ST-segment elevation with type 1 morphology of ≥2 mm in one or more of the right precordial leads V1 and V2 positioned in the second, third, or fourth intercostal space, occurring either spontaneously or after provocative drug test with intravenous administration of sodium channel blockers such as ajmaline, flecainide, procainamide or pilsicainide.1,7 Given the risk of potentially lethal ventricular arrhythmias associated with the syndrome, diagnosis is crucial, as are counseling and screening of family members, in view of the mainly autosomal dominant transmission of the mutations involved.2
The family tree generated by these three clinical cases raises important issues that need to be addressed. The family study, which began with case 3 in which a mutation associated with Brugada syndrome was identified, is a clear example of the importance of genetic testing in cases of sudden death of unexplained cause.
In up to one-third of cases of sudden death, the medico-legal autopsy finding is inconclusive, and the role of postmortem molecular autopsy is becoming increasingly important and is in fact covered in international guidelines.1,2,5,8
In a study of 49 cases of sudden death unexplained on autopsy, mean age at death of 14.2 years, mutations associated with long QT syndrome were identified in nearly 20% of cases, while mutations in the ryanodine receptor gene, associated with catecholaminergic ventricular tachycardia, were found in 14%.9 In a retrospective analysis of patients with mutations associated with long QT syndrome, 33% of those who suffered sudden death had a history of syncope and 56% had a family history of cardiac events; genetic study identified 23 relatives with the mutation, with therapeutic implications.10 Another study, on 173 patients with a mean age of 18.4±12.9 years (range 1-69 years) with autopsy-negative sudden death referred for postmortem genetic testing, analyzed mutations in genes associated with long QT syndrome and catecholaminergic ventricular tachycardia, and found pathogenic mutations in 26% of cases.10 Finally, a study on 15 patients aged between one and 35 years with non-molecular autopsy-negative sudden death assessed the presence of mutations associated with Brugada syndrome, long QT syndrome and catecholaminergic ventricular tachycardia, and identified pathogenic mutations in 40% of cases; ten first-degree family members were subsequently found to be mutation carriers.11
All these studies highlight the importance of molecular autopsy in cases of sudden death.
There are various guidelines in the literature on the clinical approach to relatives of individuals who have suffered sudden death.1,2,11,12 Several countries have implemented protocols for investigating victims of sudden death with inconclusive standard autopsy and for referring their relatives for study. The consensus statement of the Heart Rhythm Society, the European Heart Rhythm Association and the Asia Pacific Heart Rhythm Society on inherited arrhythmia syndromes recommends screening of family members in cases of unexplained sudden death, including clinical history, electrocardiography (12-lead ECG with leads V1 and V2 in the second, third and fourth intercostal spaces, 24-hour Holter monitoring, exercise testing and pharmacological provocation) and imaging studies (echocardiography and possibly cardiac magnetic resonance imaging).3 In these guidelines, genetic screening of first-degree relatives of victims of unexplained sudden death whenever a pathogenic mutation has been identified has a class I recommendation, reflecting the value of molecular study in such cases.
In Portugal, there are legal constraints on access to the results of postmortem genetic testing as part of a medico-legal autopsy. When such tests are ordered by the pathologists performing the autopsy, the results are included confidentially in the autopsy report and sent to the court overseeing the process, which must grant authorization before any third parties can access them. Only if the victim's family requests a copy of the medico-legal autopsy from the court for their own benefit or to give to the attending physician can the latter use the results as a basis for referring relatives for screening. There is thus no mechanism for making these results directly available for standardized consultations to assess relatives of victims of sudden death.
Another aspect of this case is that relatives are not assessed in cardiology consultations unless they are brought to physicians’ attention by the index case. The patient in case 2 above did not communicate the possibility of hospital screening to her relatives and thus an opportunity was lost to assess their genetic susceptibility and to identify individuals at high risk for sudden death.
Finally, with regard to the patient in case 1, we will never know for certain whether she had the mutation. Her mother also refused to undergo genetic testing, but if she had also had the mutation, this would have strengthened the possibility that her daughter had had an atypical presentation of Brugada syndrome. Left ventricular noncompaction is known to be overdiagnosed,13,14 while monomorphic ventricular tachycardia is a rare manifestation of Brugada syndrome.15–19 This, coupled with the absence of type 1 Brugada pattern on the baseline ECG, may have resulted in failure to diagnose Brugada syndrome in this individual.
ConclusionsPostmortem genetic testing of victims of sudden death with inconclusive autopsy findings is of considerable clinical value in enabling diagnosis of hereditary diseases associated with malignant ventricular arrhythmias and making it possible to prevent sudden death. To bring Portugal into line with other countries, a protocol needs to be put into place for screening of relatives of individuals who have suffered sudden death associated with hereditary diseases.
Please cite this article as: Ribeiro S, Coelho L, Puentes K, Miltenberger-Miltenyi G, Faria B, Calvo L, et al. Teste genético post mortem, o diagnóstico clínico não se esgota com a morte do doente. Rev Port Cardiol. 2019;38:503–509.
