
We describe the case of a 47-year-old man with new-onset heart failure who was found to have severe biventricular wall thickening. We present comprehensive data from invasive and non-invasive multimodality imaging, genetic and histologic tests, and briefly describe their importance in the final diagnosis.
To our knowledge, this is the first case of the Portuguese variant of familial amyloid polyneuropathy presenting with heart failure in the fifth decade of life.
This is an unusual case report, but also an illustration of how to approach any patient with suspected infiltrative cardiomyopathy.
Descrevemos o caso de um doente do sexo masculino de 47 anos, com insuficiência cardíaca de novo, que apresentava marcado aumento da espessura da parede de ambos os ventrículos. Apresentamos dados dos exames de imagem, estudo genético e análise histológica que nos guiaram no diagnóstico, explicando o nosso precurso pelos diagnósticos diferenciais e a interpretação dos diferentes testes.
Tanto quanto sabemos, é o primeiro caso descrito de um doente com a variante portuguesa da polineuropatia amiloidótica familiar que se apresenta com insuficiência cardíaca na quinta década de vida.
Este é um caso clínico invulgar, mas também um exemplo ilustrativo da abordagem de um doente com suspeita de cardiomiopatia infiltrativa.
Increased ventricular wall thickness can be a physiologic response to pressure overload or to exercise, but it can also be pathologic, resulting from myocyte hypertrophy or deposition of an abnormal substance. Finding its exact cause can be challenging, but is crucial in determining the appropriate management.
We describe the case of a 47-year-old Caucasian man complaining of tiredness and dyspnea on moderate exertion lasting several months, progressively worsening and associated with orthopnea.
A history of erectile dysfunction, night sweats and paresthesias in both hands and feet was also reported. He had also experienced bilateral calf pain during the previous month, with no specific relieving or aggravating factors, and had noted blurred vision in the left eye for the previous six months.
He denied chest pain, fever, muscular weakness or other symptoms. His medical history was positive for type 2 diabetes and his family history was unremarkable.
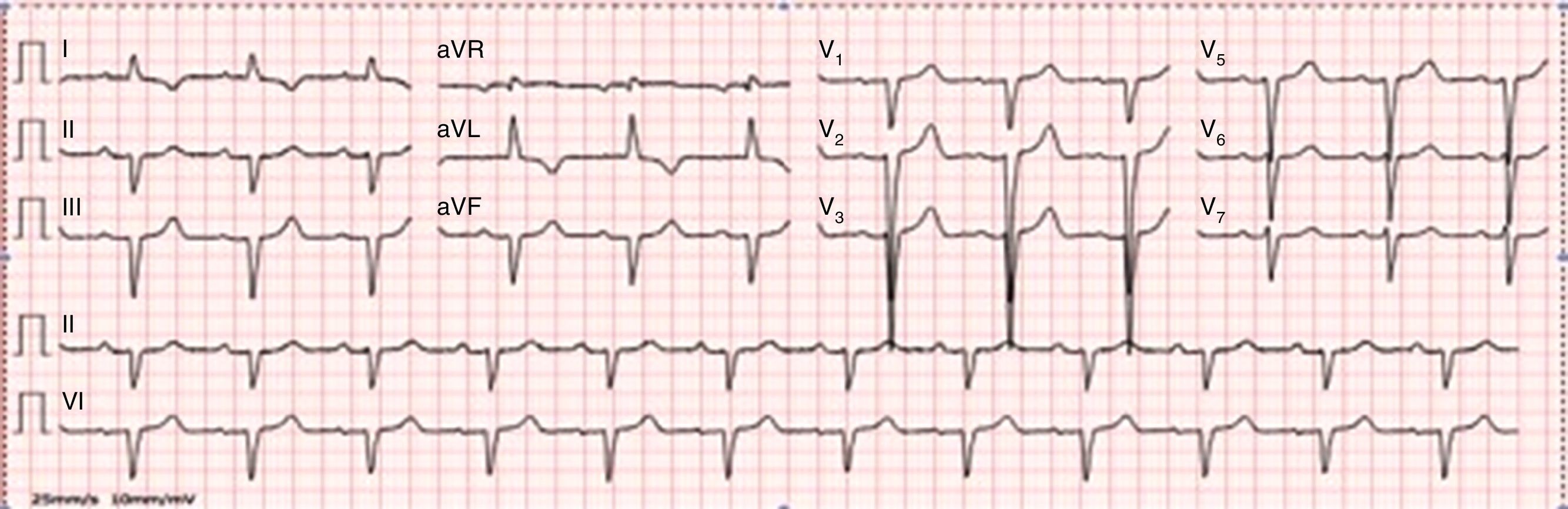
On physical examination, bilateral rales and hypoesthesia of the hands and feet were noted. The chest X-ray revealed an increased cardiothoracic index and arterial blood gas analysis showed mild hypoxemia. Blood analysis revealed increased proBNP (1781 pg/ml), without anemia or renal impairment. The electrocardiogram showed normal sinus rhythm, QS waves in the inferior leads, poor R progression in the precordial leads and T-wave inversion in I and aVL (Figure 1).

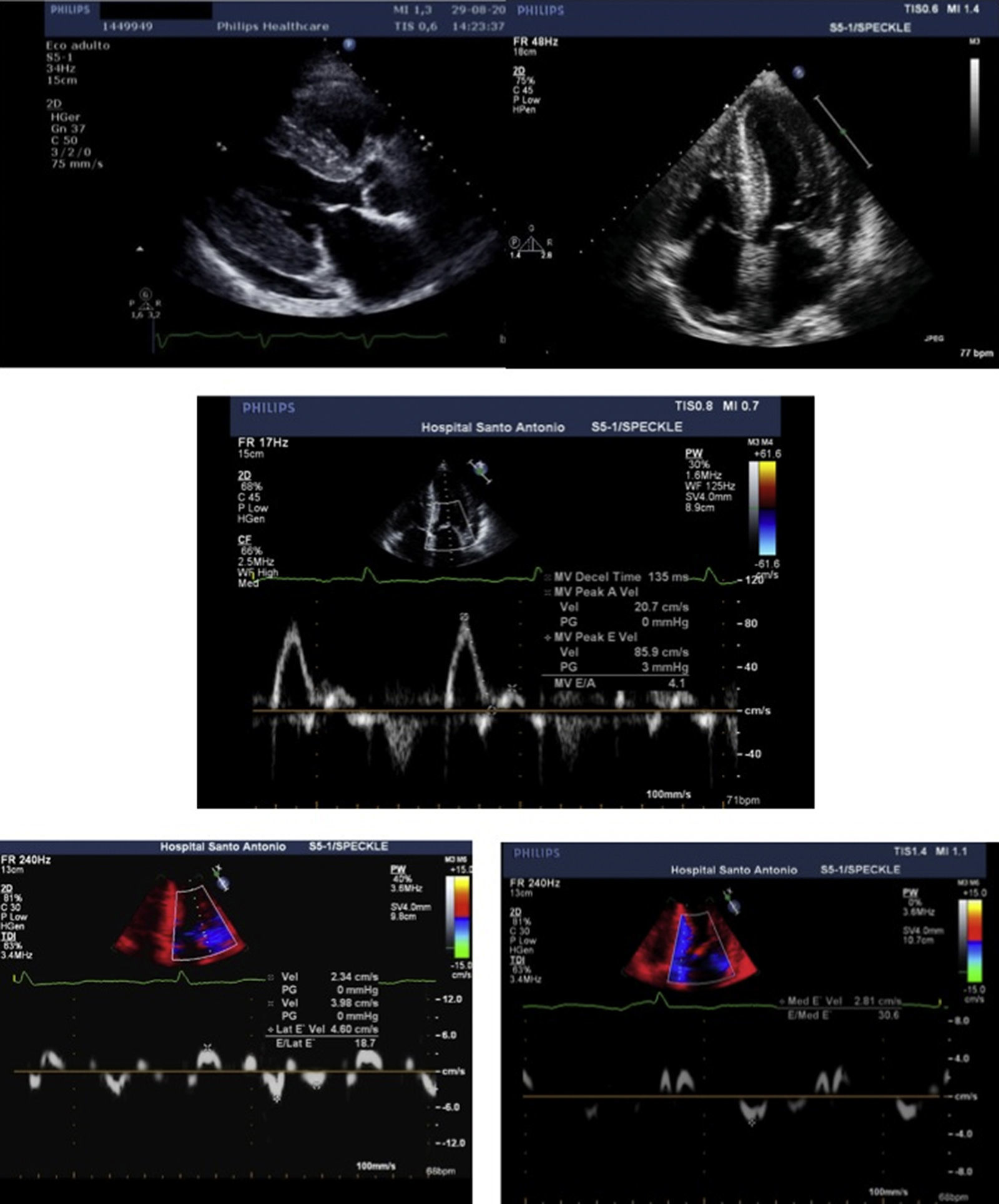
The transthoracic echocardiogram (Figure 2) showed severe biventricular concentric thickening with normal ejection fraction. Diastolic assessment indicated restrictive physiology; mild pericardial effusion was also noted.
 and 4-chamber (B) views, showing biventricular hypertrophy (interventricular septum 21 mm; posterior wall 19 mm), increased thickness of the interatrial septum, biatrial enlargement (left atrial indexed volume 42 ml/m2) and mild circumferential pericardial effusion. Diastolic assessment shows a restrictive pattern: transmitral flow (C) with E/A ratio >2 and deceleration time <150 msec; decreased tissue Doppler (D and E) E′ velocities (lateral E′: 4.6 cm/s; septal E′: 2.8 cm/s) and increased E/E′ (mean E/E′ ratio: 25). Decreased myocardial velocities on pulse-wave Doppler (lateral S′: 3.7 cm/s, septal S′: 3.3 cm/s) can also be observed (D and E).")
Transthoracic echocardiogram, parasternal long-axis (A) and 4-chamber (B) views, showing biventricular hypertrophy (interventricular septum 21 mm; posterior wall 19 mm), increased thickness of the interatrial septum, biatrial enlargement (left atrial indexed volume 42 ml/m2) and mild circumferential pericardial effusion. Diastolic assessment shows a restrictive pattern: transmitral flow (C) with E/A ratio >2 and deceleration time <150 msec; decreased tissue Doppler (D and E) E′ velocities (lateral E′: 4.6 cm/s; septal E′: 2.8 cm/s) and increased E/E′ (mean E/E′ ratio: 25). Decreased myocardial velocities on pulse-wave Doppler (lateral S′: 3.7 cm/s, septal S′: 3.3 cm/s) can also be observed (D and E).
At this point, our findings suggested an infiltrative cardiomyopathy, such as amyloidosis, Fabry disease or glycogen storage disease.1,2 The neuropathy and visual changes could fit with a systemic disease. Interestingly, the ECG did not show decreased voltages, which is common in familial amyloidosis.
Hypertrophic cardiomyopathy was another hypothesis. Against it was the ECG showing poor R-wave progression and echocardiographic findings suggestive of an infiltrative disease, with symmetric biventricular wall thickening and pericardial effusion. We excluded hypertensive heart disease, since the patient did not have hypertension. Blood analysis and thoracic computed tomography showed no changes suggestive of hemochromatosis or sarcoidosis. Thyroid disease and HIV, HCV or HBV infection were also excluded.
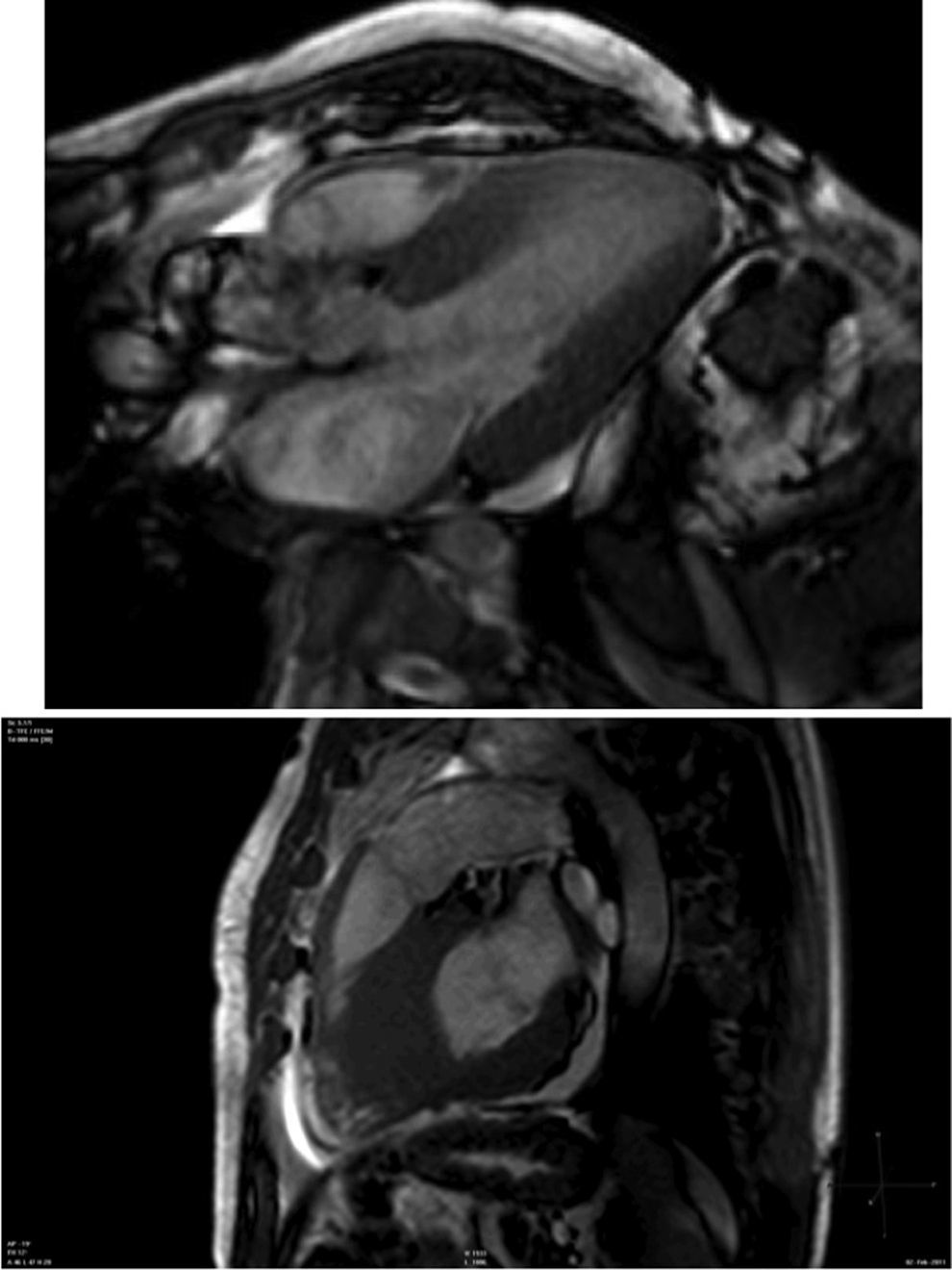
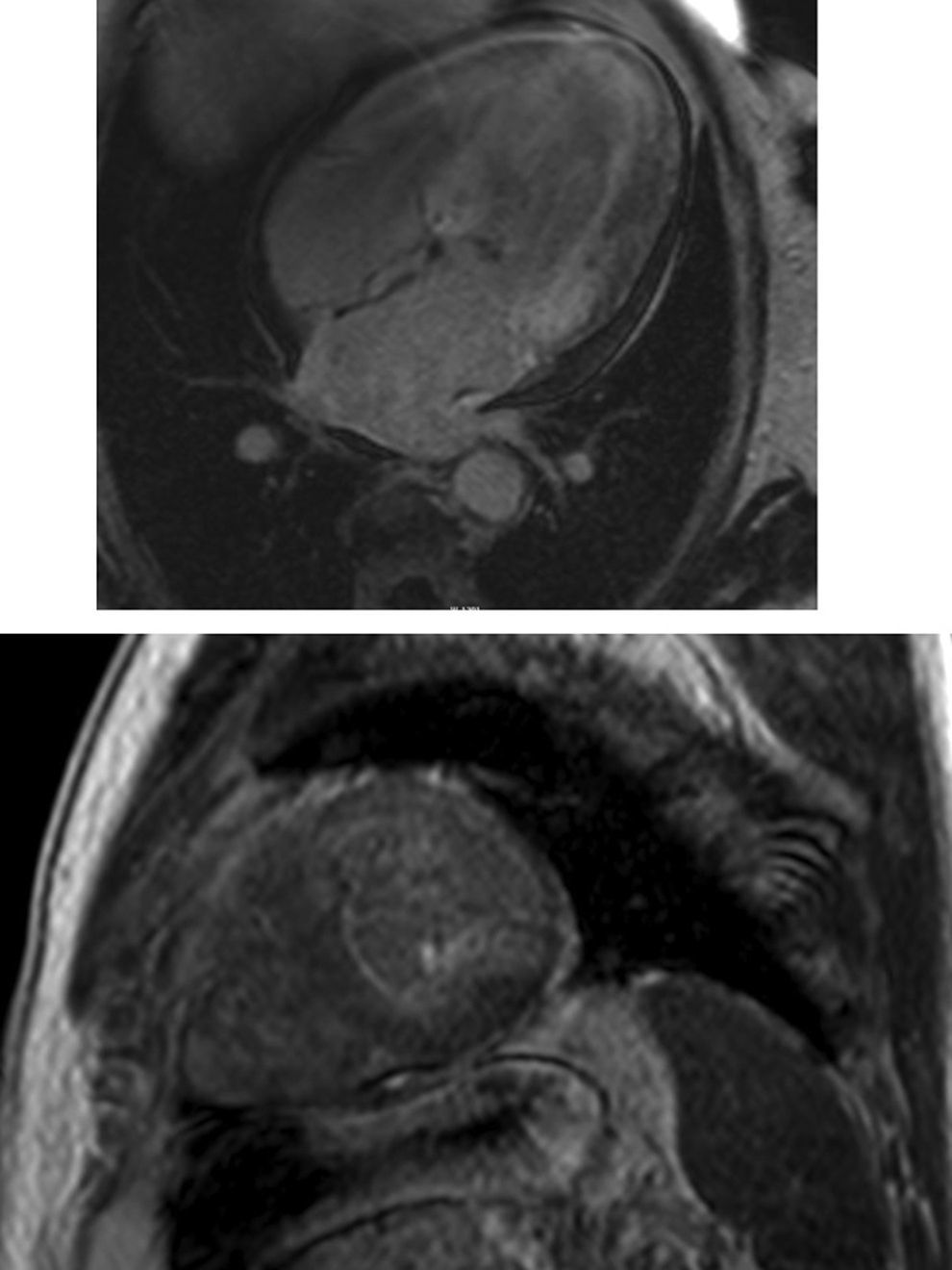
Cardiac magnetic resonance (CMR) showed marked concentric left ventricular (LV) hypertrophy, mild right ventricular (RV) hypertrophy and normal ejection fraction. There was biatrial enlargement, thickening of the interatrial septum and mild pericardial effusion (Figure 3). Short-tau inversion-recovery (STIR) sequences showed no myocardial edema or inflammation. In late gadolinium enhancement study, there was difficulty in setting the optimal inversion time to correctly null the myocardium, with a relatively dark blood pool and diffuse subendocardial enhancement of the interatrial septum and the LV and RV walls (Figure 4). The diffuse subendocardial late gadolinium enhancement, coupled with abnormal myocardial and blood-pool contrast kinetics, was most consistent with cardiac amyloidosis. The fact that the optimal inversion time that would null the normal myocardium could not be found is an indirect sign suggesting amyloidosis, because if abnormal fibrils are widespread in the intercellular space, the gadolinium may not be taken up by healthy myocardium.3


Cardiac magnetic resonance, 4-chamber view, depicting the late phase after gadolinium injection, with widespread subendocardial hyperenhancement of the left ventricle, not matching any coronary artery territory, but also involving the right ventricle, both atria and the interatrial septum.
Endomyocardial biopsies and biopsy of the salivary glands were then performed but were negative for amyloid.
Immunosubtraction testing for free immunoglobulin chains in serum and urine, which could point to AL amyloidosis, was also negative.
An electromyogram suggested the presence of a pressure-sensitive neuropathy of the lower limbs, bilateral carpal tunnel syndrome and signs of parasympathetic dysfunction, which would be compatible with amyloidosis.
Finally, we returned to the patient's complaint of left visual impairment. Vitreous opacities in the left eye were reported and, after vitrectomy, vitreous humor analysis confirmed the presence of amyloid.
Since the patient had no abnormal immunoglobulin chain production or inflammatory condition suggesting AL or AA amyloidosis, we searched for the most common mutation in Portugal of a protein responsible for the production of amyloid, and a mutation in the transthyretin gene (TTR Val30Met) was found, leading to a diagnosis of familial amyloid polyneuropathy (FAP) with a Portuguese type 1 variant.4
The patient gradually improved and was discharged after a few days. He is currently being assessed for possible heart and liver transplantation and genetic counseling has been offered to his only son.
To summarize, when assessing a patient with increased ventricular wall thickness, it is important to determine whether it is a restrictive cardiomyopathy as opposed to other types of disease, such as hypertrophic cardiomyopathy or hypertensive heart disease.5 The echocardiogram is crucial, raising the initial suspicion based on structural abnormalities and restrictive physiology. Further study with cardiac magnetic resonance and endomyocardial biopsy can help to identify an infiltrative cardiomyopathy and usually establish the diagnosis. Due to the systemic nature of these deposition diseases, it is essential to investigate the involvement of other organs and to check the family history.6
Amyloidosis consists of a group of diseases characterized by changes in secondary protein structure, in which insoluble extracellular fibril deposits form, leading to organ dysfunction.7
FAP is usually diagnosed in the third decade of life. Its predominant feature is sensorimotor neuropathy,8 but patients commonly have autonomic dysfunction, renal failure and cardiac involvement, which is the major prognostic factor.
The most common mutations affect the transthyretin gene (FAP-I or -II), but apolipoprotein A1 (FAP-III) or gelsolin (FAP-IV) may also be involved. Transthyretin is produced mainly in the liver, with a small percentage in the choroid plexus and the retina.
Amyloidosis due to a TTR mutation is an autosomal dominant disease. Many mutations have been described, but the Val30Met mutation is the most frequent mutation in Portugal, where it has more than 90% penetrance.
The cornerstone of treatment has been liver transplantation, particularly when there is already cardiac or renal impairment. However, a new drug (tafamidis) that promotes stabilization of the transthyretin protein has been approved in Europe and has shown promising results in halting progression of the disease at an early stage.
This case illustrates a highly atypical presentation of FAP, with advanced heart disease and mild neurologic impairment, relatively late onset and no family history. In fact, we believe this is the first case described so far of FAP caused by a TTR Val30Met mutation with predominantly cardiac involvement in this age-group.
This case is a reminder of the diagnostic limitations of endomyocardial biopsy and of the value of genetic testing in patients with cardiomyopathies,9 which in this instance was crucial not only to establish a definite diagnosis, but also to outline the prognosis, treatment options and follow-up for the patient and his family.
It also highlights the importance of multimodality cardiac imaging in the diagnosis of cardiomyopathies. In this case, both the echocardiogram and CMR were fundamental in guiding our investigation. A 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD) scintigraphy scan could also have helped to identify TTR amyloid deposition in the heart.
Finally, this case highlights the importance of combining all the phenotypic manifestations and persevering in determining a diagnosis, which is essential for deciding on the therapeutic approach and counseling.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe author has no conflicts of interest to declare.
