
Emery-Dreifuss muscular dystrophy type 1 (EDMD1) is a familial disease with X-linked recessive transmission, caused by a mutation in a nuclear envelope protein, emerin. Clinical manifestations usually occur in adolescence and include contractures, muscle atrophy and weakness, and cardiac conduction disturbances. We describe the case of a young male, aged 16, with first-degree atrioventricular (AV) block and limited extension of both forearms. He had elevated CK, and cardiac monitoring showed severe conduction tissue disease, with significant sinus pauses, chronotropic incompetence and periods of AV dissociation during exercise. Immunohistochemical staining using an emerin antibody showed absence of the protein in a fragment of muscle tissue and genetic study identified a mutation associated with EDMD1. Study of his brother, aged 21, also established a diagnosis of EDMD1. Both individuals received a permanent pacemaker but musculoskeletal manifestations at that time did not warrant any other intervention. Screening for certain genetic diseases, including muscular dystrophies, is mandatory following identification of conduction abnormalities in young people.
A distrofia muscular de Emery Dreifus tipo 1 (DMED1) é uma doença familiar, com transmissão recessiva ligada ao X, resultante da mutação de uma proteína do invólucro nuclear, a emerina. As manifestações clínicas ocorrem geralmente na adolescência e incluem contracturas, atrofia e fraqueza musculares e perturbações da condução cardíaca. Descreve-se o caso clínico de um jovem de sexo masculino, 16 anos, com bloqueio auriculo-ventricular (AV) de primeiro grau e limitação da extensão de ambos os antebraços. Apresentava CK elevada e a monitorização cardíaca demonstrou doença grave do tecido de condução, com pausas sinusais significativas, insuficiência cronotrópica e períodos de dissociação AV durante o esforço. A imunomarcação com anticorpos anti-emerina de um fragmento de tecido muscular confirmou a ausência desta proteína e o estudo genético identificou uma mutação associada à DMED1. O estudo do seu único irmão, de 21 anos, estabeleceu igualmente o diagnóstico de DEMD1. Por apresentarem ambos doença do tecido de condução significativa decidiu-se por implantação de pacemaker definitivo nos dois casos. As manifestações músculo-esqueléticas, de momento, não justificavam qualquer intervenção. A identificação de anomalias da condução em idades jovens obriga à exclusão de determinadas doenças genéticas, nomeadamente, distrofias musculares.
Emery-Dreifuss muscular dystrophy (EDMD) is a chronic myopathy, first described by Emery and Dreifuss in 1966.1 They identified a benign X-linked recessive form, now termed EDMD type 1 (EDMD1). Later, in the 1980s, others described EDMD with autosomal transmission2; the prevalence of both forms is poorly defined.3 They result from mutations in genes that code for nuclear envelope proteins. The main clinical manifestations include early development of contractures, progressive muscular atrophy and weakness, and cardiac conduction disturbances. Diagnostic suspicion is based on these clinical and electrocardiographic findings, which can be confirmed by muscle biopsy and genetic study.
Case reportA young male, Caucasian, aged 16, the second child of non-consanguineous parents, was referred for cardiology consultation due to electrocardiographic alterations. The only symptoms he reported were sporadic dizziness and fatigue during sports activities, but no pre-syncope, syncope or palpitations. His personal history included previous diagnoses of allergic asthma and rhinitis. The only relevant family history was the premature death of a maternal aunt, probably due to neuromuscular disease. Physical examination revealed uncharacteristic facial features, normal body mass index, irregular heartbeat on cardiac auscultation but no murmur, normal pulmonary auscultation, palpable and symmetrical radial and femoral pulses, and soft abdomen, with no organomegaly.
Routine laboratory tests revealed no abnormalities, except for creatine kinase (CK) of 887UI/l, five times the normal upper limit (<171UI/l). Thyroid hormone levels were normal.
The electrocardiogram (ECG) showed sinus rhythm, first-degree atrioventricular block (AVB), 70° electrical axis, and normal QRS duration and corrected QT interval.
Further cardiac study, including Holter ECG monitoring, showed sinus rhythm, with minimum, mean and maximum heart rates of 32, 54 and 90bpm, respectively, and periods of first-degree AVB. Frequent supraventricular extrasystoles were also detected, with episodes of supraventricular tachycardia of three complexes, infrequent ventricular extrasystoles and multiple sinus pauses, the longest lasting 11020ms, at 00.50am, asymptomatic. The echocardiogram showed normal-sized chambers, good biventricular function and no valve abnormalities. Exercise testing revealed marked chronotropic incompetence, only 54% of maximum heart rate being attained after 12min, with periods of atrioventricular dissociation at peak exercise. Electrophysiological study documented sinus node dysfunction, with prolonged recovery time (5280ms).
Significant conduction defects associated with moderate CK elevation suggested neuromuscular disease with cardiac involvement. When questioned about musculoskeletal symptoms, the patient reported slight limitation of elbow extension only, previously disregarded since it did not significantly limit his functional capacity.
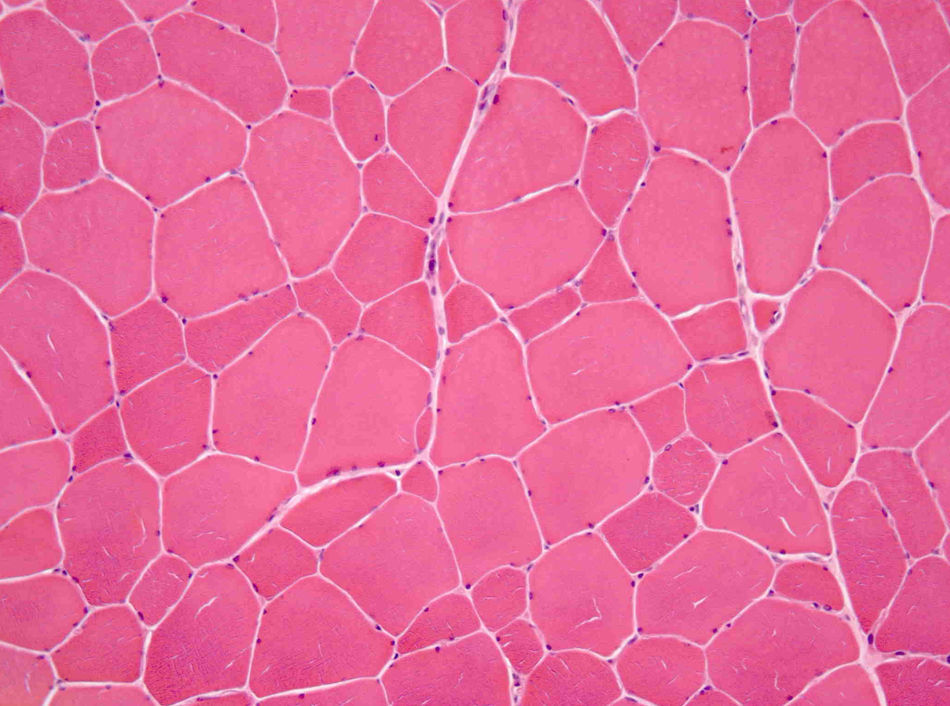
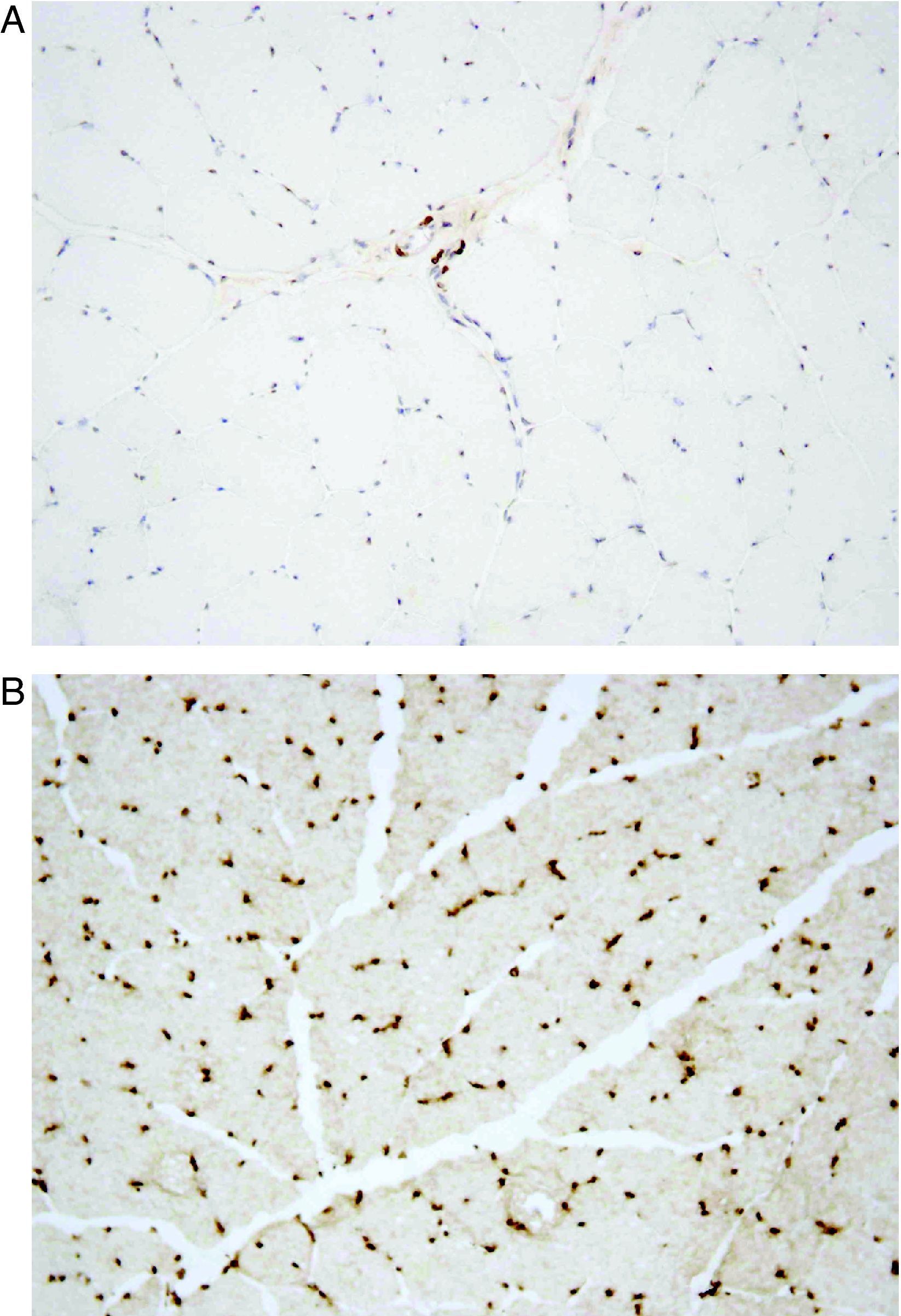
He was subsequently referred to our hospital for a neuromuscular disease consultation. Physical examination revealed moderate contractures of both elbows, with no muscle atrophy or weakness or sensory or motor deficit. Needle electromyography of the deltoid muscles and brachial biceps was normal, as was the study of sensory and motor nerve conduction. Biopsy of the left deltoid muscle revealed increased variation in muscle fiber diameter (Fig. 1), and immunohistochemical analysis using an emerin antibody showed absence of immunostaining in all muscle fiber nuclei (Fig. 2A and B). Genetic study confirmed the diagnosis of EDMD1 by identifying the mutation c.266-27_266-10del in the EMD gene.
.")
 compared to control (B); 200×.")
Once the diagnosis was established, it was decided to implant a permanent pacemaker (DDDR). The skeletal contractures did not warrant any other intervention.
Assessment of the patient's brother, aged 21, revealed an irregular heart beat on cardiac auscultation, and neurological study identified myotatic hyporeflexia, mild atrophy, no upper limb muscle weakness, forearm extension limited to +150/160° and preserved spinal column mobility. His CK level was also elevated (613UI/l). The electrocardiogram revealed atrial fibrillation with controlled ventricular rate, but no other alterations. Holter 24h monitoring confirmed this basic rhythm and a mean heart rate of 60bpm. Infrequent ventricular extrasystoles were also detected, as well as multiple sinus pauses, the longest lasting 3300ms. Normal blood pressure and heart rate changes were recorded during exercise testing, with no AV conduction disturbances. No structural heart disease was detected on echocardiography. After genetic study identified the same mutation as found in his brother and established a diagnosis of EDMD1, it was decided to implant a permanent pacemaker (VVIR).
The two brothers are being monitored in annual consultations in cardiology and neurology. They remain asymptomatic from a cardiac standpoint, with no musculoskeletal signs of disease progression.
DiscussionEmery-Dreifuss muscular dystrophy type 1 results from a mutation in the EMD gene coding for the protein emerin, at locus Xq28. This protein is found in the inner nuclear membrane of virtually all cells, but is expressed more in the nuclei of skeletal and cardiac muscle cells. Its functions are poorly understood, but it is assumed to play a crucial role in regulating gene expression and maintaining nuclear structure. More than 70 mutations are currently known, most of which result in the complete loss of emerin, although in some cases reduced production is observed.4 There is no correlation between the genetic defect and phenotypic expression, as reflected in the enormous clinical variability, not only between affected families but also within the same family.5 The cases presented illustrate this point, since the two brothers were carriers of the same mutation but developed different cardiac manifestations. The fact that both sons of apparently healthy non-consanguineous parents were affected suggests X-linked recessive transmission, in which the mother must be a carrier. Further evidence for this form of transmission is the fact that a maternal aunt probably died of neuromuscular disease.
Autosomal forms have similar clinical expression, but result from mutations in the LMNA gene that codes for lamins A and C, filament proteins found in the inner nuclear membrane and nucleoplasm of almost all cells that provide structural support and regulate DNA replication.
In X-linked EDMD, symptoms generally appear in adolescence, but manifestations of the disease can occur at any time between the neonatal period and the third decade of life.6 The classical clinical triad includes contractures, most commonly of the elbows, Achilles tendons, and postcervical muscles; muscle wasting and weakness with a humeroperoneal distribution in the early stages; and cardiac conduction disturbances.
Contractures usually precede the development of muscle atrophy7 but rarely lead to complete loss of mobility.8 Prompt rehabilitation or surgical correction can minimize functional impairment.
Muscle weakness and atrophy usually have a symmetrical bilateral distribution and progress slowly. The proximal upper and distal lower limb muscles are most commonly affected in the initial stages, while the pectoral and pelvic girdles are generally affected later.
Anomalies in the formation and conduction of cardiac electrical stimuli are common, to the extent that it is rare for affected individuals to present a normal ECG after the age of 35–40.9 Such alterations usually appear after development of muscle weakness and can cause syncope or even sudden death. The latter may in fact be the first manifestation, hence the difficulty in determining the actual prevalence of the disease.6 Many abnormalities have been described, including sinus bradycardia, supraventricular and ventricular arrhythmias and AV block. PR interval prolongation is a common initial finding.8 The atria are more frequently affected than the ventricles and thus supraventricular arrhythmias such as fibrillation and flutter are common.9 One typical finding is junctional rhythm associated with atrial standstill. In more advanced stages, myocytes may be replaced by fibroadipose tissue,10 leading to contractile dysfunction and cardiac chamber dilatation.
In the cases presented, muscle abnormalities also preceded cardiac manifestations, but had been disregarded, since they did not significantly limit functional capacity. The associated cardiac conduction disturbances justified implantation of a permanent pacemaker in both cases, whereas the musculoskeletal manifestations were mild and required no treatment.
Prompt diagnosis of EDMD is important; the condition should be suspected following the clinical findings described above. Slight CK elevation, present in both our patients, strengthens the suspicion of muscular disease. Marked elevation (more than ten times the normal upper limit) suggests other types of dystrophy, particularly Duchenne or Becker.7 Normal CK values are rare, but do not exclude the diagnosis11. ECG is recommended in all patients,6 and echocardiography should be performed to exclude structural heart disease. The electromyogram may be normal or show various alterations (increased insertional activity, fibrillation, positive waves), which help in confirming the myopathic nature of the disease and differential diagnosis.12 A muscle biopsy should be performed in all cases of suspected EDMD1. The most common histological finding is variation in muscle fiber diameter,11 as found in our case. Necrotic and regenerative fibers and increased endomysial connective tissue may also be detected. Use of complementary immunohistochemical techniques facilitates differential diagnosis with other myopathies.13
There is no specific treatment for EDMD1. Permanent pacemaker implantation is recommended for all patients with evidence of conduction tissue disease,9 as it reduces the risk of sudden death,11,14 although there have been reports of sudden death even after pacemaker implantation.9 At present, there are no clear indications for prophylactic implantation of a cardioverter-defibrillator.9 A non-randomized study demonstrated a significant percentage of appropriate shocks, which supports implantation,15 but additional studies are required to establish clear guidelines.
In complicated cases, with development of dilated cardiomyopathy, standard heart failure therapy is recommended. Heart transplantation is justified in refractory cases.
The clinical course is usually benign, despite the possibility of severe contractures and the risk of sudden death. While female carriers do not develop musculoskeletal symptoms, they can have conduction disorders, and there have been some reports of sudden death.9 An annual ECG is therefore recommended.6 The ECG of the mother in the cases presented was normal.
Before treating conduction disturbances in young individuals, certain rare genetic diseases should be excluded, particularly muscular dystrophies that are associated with cardiac involvement.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Saraiva, F, et al. Distrofia muscular de Emery-Dreiffus: a propósito de um caso clínico. Rev Port Cardiol. 2012. doi:10.1016/j.repc.2012.01.006.
