





The importance of sodium channels for the normal electrical activity of the heart is emphasized by the fact that mutations (inherited or de novo) in genes that encode for these channels or their associated proteins cause arrhythmogenic syndromes such as the Brugada syndrome and the long QT syndrome (LQTS). The aim of this study is to conduct a review of the literature on the mutations in the sodium channel complex responsible for heart disease and the implications of a close relationship between genetics and the clinical aspects of the main cardiac channelopathies, namely at the level of diagnosis, risk stratification, prognosis, screening of family members and treatment.
MethodsThe online Pubmed® database was used to search for articles published in this field in indexed journals. The MeSH database was used to define the following query: “Mutation [Mesh] AND Sodium Channels [Mesh] AND Heart Diseases [Mesh]”, and articles published in the last 15 years, written in English or Portuguese and referring to research in human beings were included.
ConclusionsIn the past few years, significant advances have been made to clarify the genetic and molecular basis of these syndromes. A greater understanding of the underlying pathophysiological mechanisms showed the importance of the relationship between genotype and phenotype and led to progress in the clinical approach to these patients. However, it is still necessary to improve diagnostic capacity, optimize risk stratification, and develop new specific treatments according to the genotype-phenotype binomial.
A importância dos canais de sódio para a normal atividade elétrica do coração é enfatizada pelo facto de as mutações (hereditárias ou de novo) nos genes que codificam esses canais ou as proteínas a esses associadas provocarem síndromes arritmogénicas como a síndrome de Brugada e a síndrome do QT longo. O objetivo deste trabalho é proceder a uma revisão bibliográfica sobre as mutações no complexo dos canais de sódio responsáveis por doença cardíaca e as implicações da relação estrita entre a genética e a clínica das principais canalopatias cardíacas, nomeadamente no nível do diagnóstico, da estratificação do risco, do prognóstico, do rastreio de parentes e terapêutica.
MétodosFoi usada a base de dados online Pubmed® para pesquisar os artigos publicados nessa área, em revistas indexadas. Recorreu-se à MeSH Database para definir a seguinte query: “Mutation [Mesh] AND Sodium Channels [Mesh] AND Heart Diseases [Mesh]” e incluíram-se artigos publicados nos últimos 15 anos, escritos em inglês ou português e referentes à investigação em humanos.
ConclusõesNos últimos anos, grandes avanços foram feitos no esclarecimento da base genética e molecular dessas síndromes. A maior compreensão dos mecanismos fisiopatológicos subjacentes demonstrou a importância da relação entre o genótipo e o fenótipo e permitiu efetuar progressos na abordagem clínica desses pacientes. Todavia, é ainda necessário melhorar a capacidade de diagnóstico, aprimorar a estratificação do risco e desenvolver novas terapêuticas específicas de acordo com o binómio genótipo-fenótipo.
atrial fibrillation
action potential
Brugada syndrome
calcium ion
caveolin-3
cardiac conduction disease
catecholaminergic polymorphic ventricular tachycardia
dilated cardiomyopathy
electrocardiogram
electrophysiological study
heart rate
calcium current
potassium current
sodium current
late, persistent or sustained sodium current
peak sodium current
potassium ion
long QT syndrome
long QT syndrome type 1
long QT syndrome type 2
long QT syndrome type 3
milliseconds
sodium ion
sodium channels
programmed ventricular stimulation
polymorphic ventricular tachycardia
QT interval corrected for heart rate
sudden cardiac death
sudden death
single nucleotide polymorphism
syntrophin
short QT syndrome
Torsade de pointes
ventricular fibrillation
ventricular tachycardia
Cardiac channelopathies constitute a heterogeneous group of inherited cardiac diseases caused by mutations in genes that encode for the ion channels expressed in the heart (involved in Na+ [INa], K+ [IK] and Ca2+ [ICa] currents) and/or the proteins that regulate their function.1–3 These mutations result in different phenotypes according to the abnormalities induced in the sodium current and in other ion currents, leading to a greater likelihood of occurrence of syncope, seizures and arrhythmias, although most of the time there are no underlying structural heart defects.4 This shows the importance of ion channels, namely sodium channels (NaC), in the genesis and propagation of the action potential (AP), and consequently in heart excitability.2,3,5–7
Induced arrhythmias are potentially fatal, and sudden cardiac death (SCD) frequently constitutes the first manifestation of these diseases.4,8 Sudden death (SD) is one of the most common causes of death due to cardiovascular pathologies and, in the adult Western population, cardiac channelopathies (1-2%) are one of the most frequently diagnosed predisposing pathologies together with cardiomyopathies (10-15%) and coronary disease (75%).9 In reality, some studies show that cardiac channelopathies are responsible for approximately 1/3 of the SD cases in young people with a negative autopsy, and up to 50% of the cases of arrhythmic SCD.10,11
The main hereditary arrhythmias caused by ion channel dysfunctions are Brugada syndrome (BrS), long QT syndrome (LQTS), short QT syndrome (SQTS) and catecholaminergic polymorphic ventricular tachycardia (PVT).4,12 However, their prevalence in the general population is difficult to estimate.11,13–15 In addition to the pathologies mentioned above, pre-excitation syndrome, idiopathic ventricular fibrillation (VF) and rare cases of familial cardiomyopathies are also associated with ion channel mutations.4,12
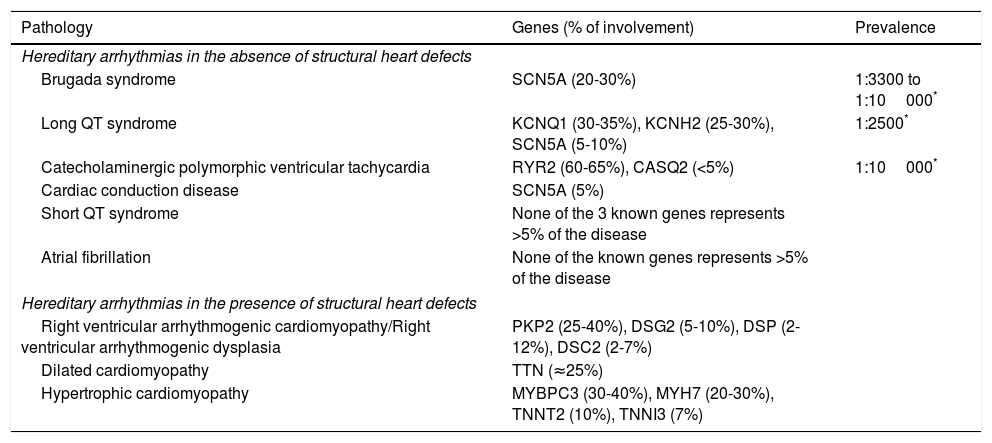
In the last two decades, the knowledge about the genetic and molecular mechanisms underlying arrhythmias (especially those of hereditary nature – Table 1) has vastly increased, and various mutations and/or genetic variants have been described.16,17
Main genes associated with hereditary arrhythmias.
| Pathology | Genes (% of involvement) | Prevalence |
|---|---|---|
| Hereditary arrhythmias in the absence of structural heart defects | ||
| Brugada syndrome | SCN5A (20-30%) | 1:3300 to 1:10000* |
| Long QT syndrome | KCNQ1 (30-35%), KCNH2 (25-30%), SCN5A (5-10%) | 1:2500* |
| Catecholaminergic polymorphic ventricular tachycardia | RYR2 (60-65%), CASQ2 (<5%) | 1:10000* |
| Cardiac conduction disease | SCN5A (5%) | |
| Short QT syndrome | None of the 3 known genes represents >5% of the disease | |
| Atrial fibrillation | None of the known genes represents >5% of the disease | |
| Hereditary arrhythmias in the presence of structural heart defects | ||
| Right ventricular arrhythmogenic cardiomyopathy/Right ventricular arrhythmogenic dysplasia | PKP2 (25-40%), DSG2 (5-10%), DSP (2-12%), DSC2 (2-7%) | |
| Dilated cardiomyopathy | TTN (≈25%) | |
| Hypertrophic cardiomyopathy | MYBPC3 (30-40%), MYH7 (20-30%), TNNT2 (10%), TNNI3 (7%) | |
Although many mutations in different ion channels affect the heart's electrical currents, we only cover what concerns sodium currents in this literature review. In particular, we cover the structure of the NaC and their role in heart excitability, mutations in the NaC complex, the associated phenotypes and the implications of the relationship between genetic and clinical aspects at the level of diagnosis, risk stratification, prognosis and treatment, namely of LQTS and BrS.
MethodsA narrative review of the literature covering the topic Cardiac channelopathies: the role of sodium channel mutations was conducted. The online Pubmed® database was used to search for articles published in this field, and the MeSH database was used to select the MeSH terms and to define the following query: “Mutation [Mesh] AND Sodium Channels [Mesh] AND Heart Diseases [Mesh]”.
Applying the predefined inclusion criteria, only articles published in the last 15 years, written in English or in Portuguese, and referring to research in human beings were included. Additionally, the impact factor was taken into consideration.
Other references were also included, some with a publication date prior to 2002, with the aim of widening the relevant content cited in the initially searched articles.
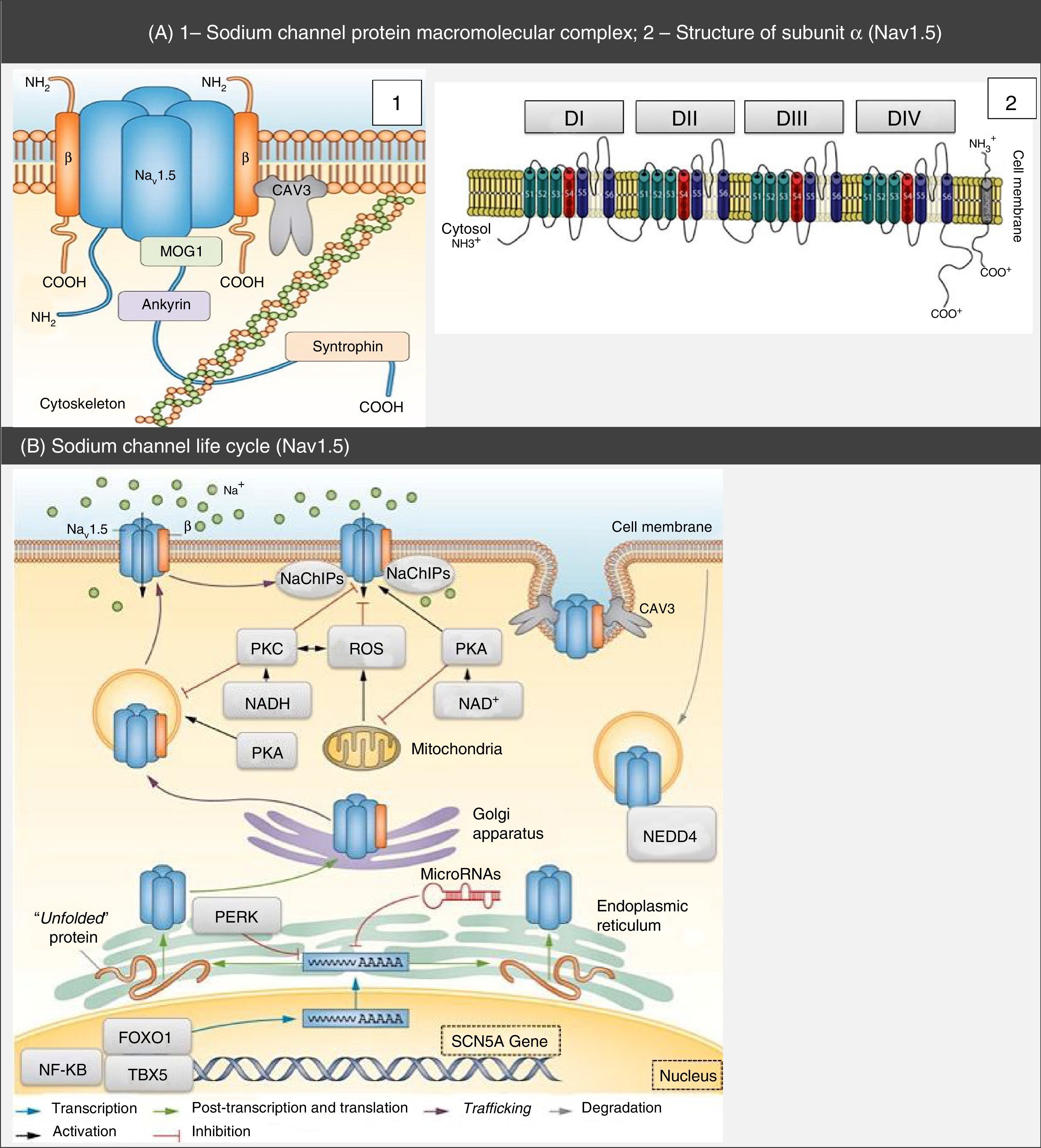
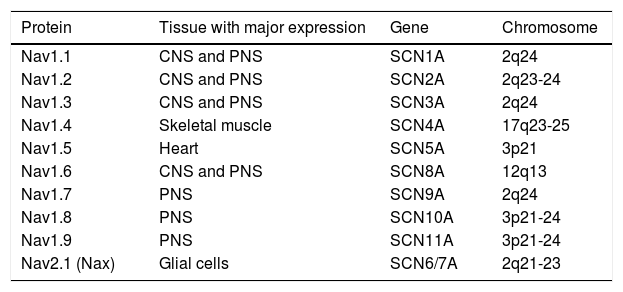
Structure and function of sodium channelsThe NaC are transmembrane proteins consisting of an α subunit together with one or two β subunits (Figure 1).2 There are various types of α subunits, which are differentially expressed according to the type of tissue and are encoded by a family of 10 different genes (Table 2).18,19 The main α subunit expressed in the heart is called Nav1.5 (like the sodium channel it is part of) and is encoded by the SCN5A (sodium channel, voltage gated, type V alpha subunit) gene, which includes 28 exons and spans more than 100 kb in chromosome 3p22.18,20
 CAV3: caveolin-3; FOXO1: forkhead box protein O1; MOG1: Ran guanine nucleotide release factor;
CAV3: caveolin-3; FOXO1: forkhead box protein O1; MOG1: Ran guanine nucleotide release factor; Protein macromolecular complex, α subunits and life cycle of the Nav1.5 sodium channels. (A) The Nav1.5 channel is part of a macromolecular complex interacting with various proteins such as: β subunits, caveolin-3, MOG1, ankyrin, syntrophin and cytoskeleton. Extracted and adapted from Liu et al. (2014)7 and Amin et al. (2010).22(B) The life cycle of Nav1.5 starts in the nucleus, where transcription of the SCN5A gene and the respective regulation by transcription factors (FOXO1, NF-KB and TBX5) occur. However, microRNAs also regulate mRNA levels. In the endoplasmic reticulum proteins are translated and, after appropriate protein folding and assembly, they are transported to the cell membrane (trafficking). Mutations or splicing variants may lead to a misfolded Nav1.5 protein, which may activate the PERK pathway for downregulation of their mRNA levels. PKA, PKC, oxidative stress (ROS) and metabolic states (NADH and NAD+) may modulate channel trafficking. NEDD4 regulates ubiquitin-mediated degradation. Extracted and adapted from Liu et al. (2014).7
CAV3: caveolin-3; FOXO1: forkhead box protein O1; MOG1: Ran guanine nucleotide release factor; NaChIP: Na+-channel-interacting protein; NEDD4: E3 ubiquitin-protein ligase NEDD4; NF-κB: nuclear factor NF-κB; PERK: eukaryotic translation initiation factor 2α-kinase 3; PKA: AMPc-dependent protein kinase (protein kinase A); PKC: protein kinase C; ROS: reactive oxygen species; TBX5: T-box transcription factor TBX5.
Sodium channel α subunits.
| Protein | Tissue with major expression | Gene | Chromosome |
|---|---|---|---|
| Nav1.1 | CNS and PNS | SCN1A | 2q24 |
| Nav1.2 | CNS and PNS | SCN2A | 2q23-24 |
| Nav1.3 | CNS and PNS | SCN3A | 2q24 |
| Nav1.4 | Skeletal muscle | SCN4A | 17q23-25 |
| Nav1.5 | Heart | SCN5A | 3p21 |
| Nav1.6 | CNS and PNS | SCN8A | 12q13 |
| Nav1.7 | PNS | SCN9A | 2q24 |
| Nav1.8 | PNS | SCN10A | 3p21-24 |
| Nav1.9 | PNS | SCN11A | 3p21-24 |
| Nav2.1 (Nax) | Glial cells | SCN6/7A | 2q21-23 |
CNS: central nervous system; PNS: peripheral nervous system.
Extracted and adapted from England and Groot (2009).19
Regulation of transcription of the SCN5A gene is influenced by many factors, including: presence of three promoters, transcription factors and microRNAs with post-transcriptional activity. More than 10 isoforms resulting from splicing of this gene have been described, and the most abundant isoform in the human heart is SCN5A-003 (adult isoform).7,18,20
The α subunit has about 227 kDa and consists of a transmembrane protein with four homologous domains (DI-DIV) connected by cytoplasmic loops, each of them with six α-helix transmembrane segments (S1-6) connected by intra- and extracellular loops. It also has a C terminus (carboxy) and an N terminus (amino), both being cytoplasmic.5,6,21
The central pore is formed by the four S5 and S6 segments of subunit α, namely by the extracellular loops that connect them. It is selectively permeable to sodium, which travels through it according to the electrochemical gradient. Segments S1 to S4 act as voltage sensors. However, the latter has the peculiarity of having a positive charge.2,5,18,21
Like the other voltage-gated channels, the NaC show conformational changes during a process called gating that enables defining three functional states for the channel (open, inactive or closed), according to membrane potential. These alterations occur in the α subunit, which is the main one responsible for regulation of depolarization of excitable cells membranes.2,18,22
The subunits are proteins of approximately 30-40 kDa, with a single transmembrane segment, an intracellular C terminus and an extracellular N terminus.5 These subunits associate with the α subunit of the NaC (Figure 1), thus not only modulating their expression on the cell surface and the gating process, but also enabling connection with the cytoskeleton and other interaction proteins. In effect, the subunits are capable of increasing the channels traffic to the cell membrane, with subsequent increase in INa.5,18
There are four types of β subunits (β1, β2, β3 and β4), encoded by the SCN1B, SCN2B, SCN3B and SCN4B genes, respectively. These are preferentially associated with different α subunits according to the type of tissue where they are expressed.5,18,20,23
In addition to the β subunits, there are other proteins with the ability to interfere and modulate Nav1.5 function (ankyrin-G, calmodulin, caveolin-3, syntrophin α1, plakophilin-2, Ran guanine nucleotide release factor [MOG1], glycerol-3-phosphate dehydrogenase 1-like [GPD1L], fibroblast growth factor homologous factor 1B [FHF-1B] and Nedd4-like ubiquitin ligases, among others) that integrate a macromolecular complex (Figure 1).5,6,18,20,21,24,25
The role of sodium channels in heart excitabilityThe heart AP is generated by depolarizing (INa; ICa) and re-polarizing (IK) ion currents.22 NaC play an essential role in AP initiation through the generation of INa, and they are expressed in the membrane of atrial and ventricular cardiomyocytes and in specialized conduction tissue.21–23 However, although their expression is abundant in the bundle of His, bundle branches and Purkinje fibers, their expression is low or absent in the sinus and atrioventricular nodes.6,21
In the ventricular myocardium, during diastole, the transmembrane electrical potential (at rest) is approximately -85 mV, and the NaC are closed. When a stimulus depolarizes the membrane, the S4 segments of the four domains move simultaneously outside, the channel opens and there is Na+ movement to the intracellular medium, according to the electrochemical gradient.5,6,18,22 In addition, INa, the main agent responsible for the rapid AP depolarization phase (phase 0), is thus generated and then rapidly increases until it reaches its peak (INapeak) and decreases milliseconds later.18,23
In NaC inactivation, the loop between domains III and IV (inactivation gate) works as a “lid” and the channels gradually close in about 1 ms.18,26 It should be noted that the NaC undergo various conformational changes that translate into different inactivation states (rapid, intermediate and slow inactivation) which, in turn, have different recovery times.18,22 However, at the end of phase 0, the majority (≈99%) of the NaC are inactivated, precluding ion traffic. They remain like this until the cell membrane is repolarized, when they recover from inactivation and again become available to be activated during phase 4.5,18
Nevertheless, during AP phase 2, a small fraction of the NaC (<1% of the total NaC available) may maintain conductibility for Na+ and reopen, and thus a small INa called late current persists (INalate).5,18,27 Moreover, some channels may reactivate during the repolarization phase (phase 3), when inactivation is not yet completed but the AP enables their reactivation, generating a current called window current.18,22 This current corresponds to less than 1% of the sodium current peak.22 An important role in the ventricular arrhythmogenesis present in certain cardiac pathologies, some of them covered in this review, has been attributed to these two currents.5,18,22,27
Under physiological conditions, the NaC activation and inactivation processes are strictly regulated in order to ensure normal cardiac electrical activity. Anomalies in the NaC cause significant abnormalities in heart electrophysiology and potentiate arrhythmogenesis, which may result from alterations in gating properties or in INa kinetics. These alterations change channel availability, the amplitude of the INapeak or prevent adequate channel inactivation, with maintenance of a persistent INa during the AP plateau. Therefore, the importance of the NaC in heart excitability is emphasized by the occurrence of potentially fatal arrhythmias (e.g., ventricular tachycardia and VF) in the presence of hereditary or acquired dysfunction in these channels.18,21,22
Mutations in sodium channelsMutations in subunit αIn the last few decades, the knowledge about the function of the SNC5A gene at the molecular and electrophysiological level has greatly increased, and various genetic studies show that mutations in this gene are associated with many heart diseases, namely hereditary cardiac arrhythmias.1,5,16,18,21,22,28 In most cases, the pathologies associated with NaC mutations are caused by mutations that alter channel permeability or the gating process.6,21
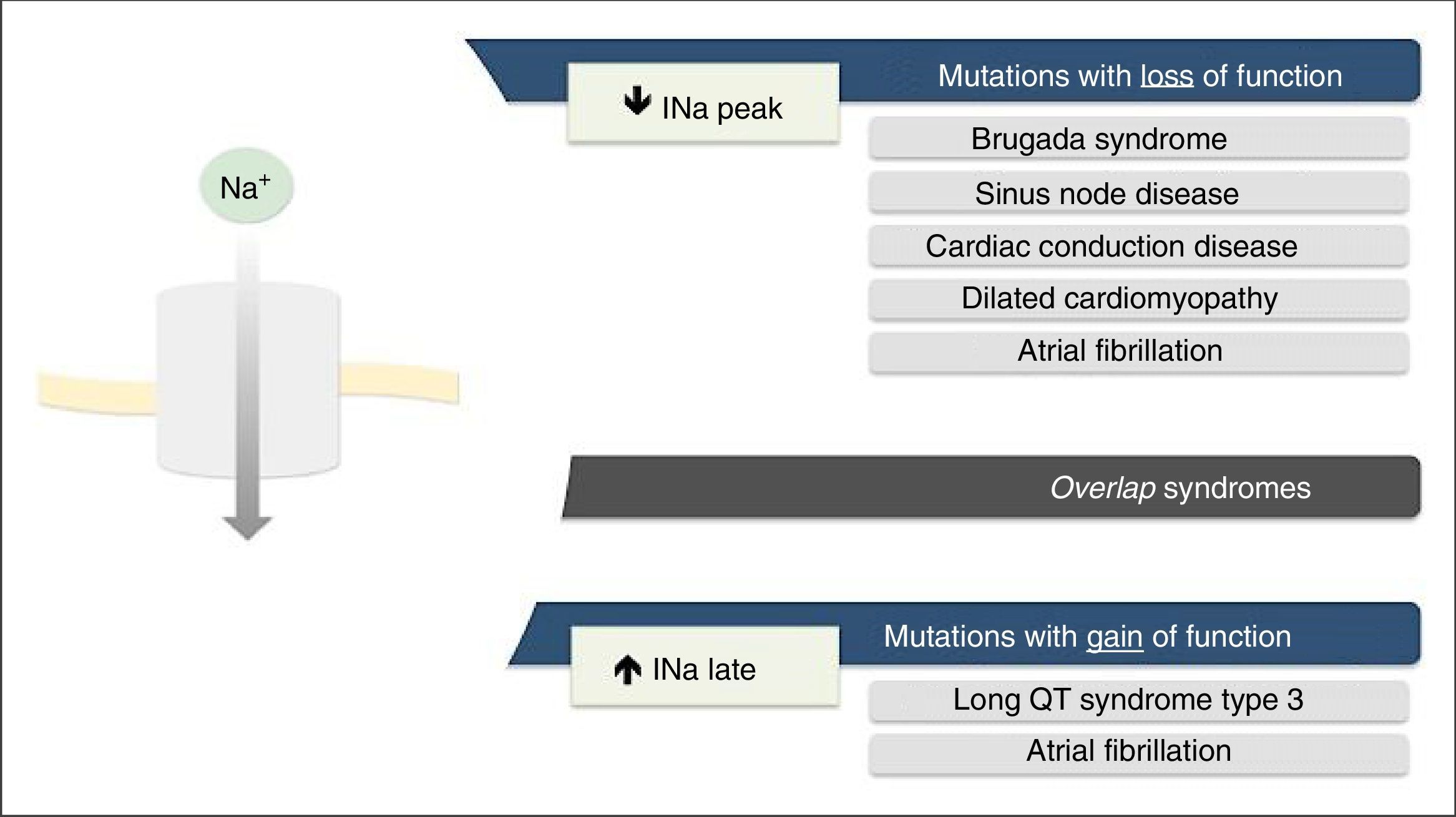
Mutations in the SCN5A gene leading to dysfunction of the Nav1.5 NaC may be due to gain of function, loss of function or both.7,18
The loss-of-function mutations result in decreased INa and are associated with BrS, sinus node disease (SND), atrial fibrillation (AF), Lev-Lenégre disease and dilated cardiomyopathy (DCM) (Figure 2).7,18 The mechanism most frequently involved is decreased INapeak (Figure 3).7,18,23
.7")
Clinical phenotypes associated with mutations in Nav1.5 sodium channels.
Extracted and adapted from Liu et al. (2014).7
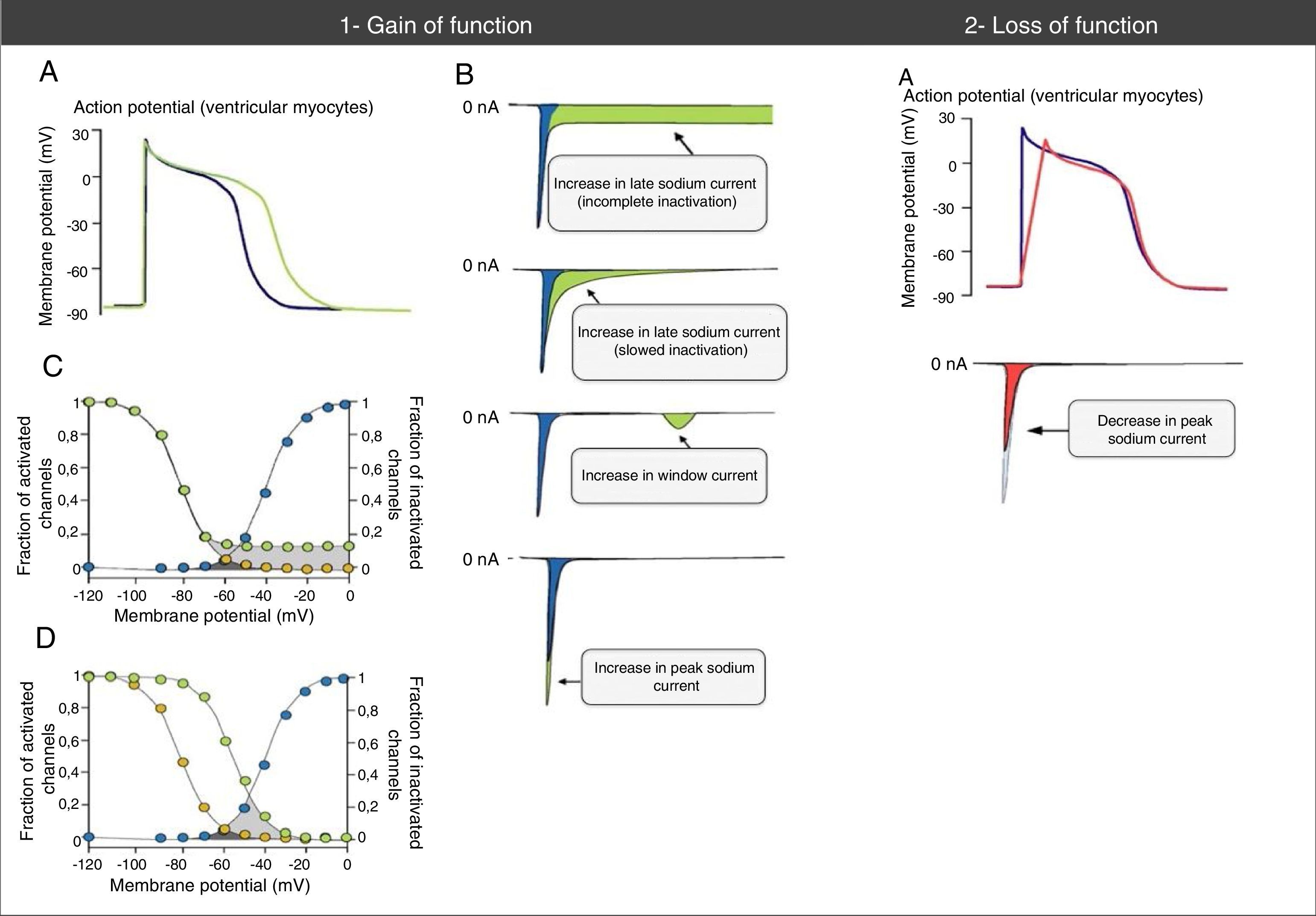
 INa during the AP phase 2 with prolonged membrane depolarization and delayed repolarization), which may be due to incomplete or slowed inactivation. Other less common mechanisms are increased window current and increased
INa during the AP phase 2 with prolonged membrane depolarization and delayed repolarization), which may be due to incomplete or slowed inactivation. Other less common mechanisms are increased window current and increased Abnormalities in action potential and sodium currents associated with gain- and loss-of-function mutations in Nav1.5 channels. (1A) Gain-of-function mutations are associated with an increase in the duration of the action potential and may trigger arrhythmic events. (1B) Various mechanisms may be associated with gain of function in the sodium current. The most common mechanism is increased late sodium current (abnormally sustained increase of INa during the AP phase 2 with prolonged membrane depolarization and delayed repolarization), which may be due to incomplete or slowed inactivation. Other less common mechanisms are increased window current and increased INa peak (increase in influx of Na+ in AP phase 0). (1C) The mechanism of sodium current increase more frequently results from incomplete inactivation of the sodium channels (green circles). (1D) In the mechanism of window current increase (green circles), inactivation occurs in more positive AP states, “delaying” and broadening the amplitude of voltages during which NaC may be reactivated. (2A) In loss-of-function mutations, decreased peak sodium current decreases the upstroke speed of the AP phase 0, slowing down electrical conduction in the heart.
Extracted and adapted from Amin et al. (2010).22
Gain-of-function mutations result in increased INa and are associated with LQTS3 (Figure 2). There are also some gain-of-function mutations associated with AF and DCM.7,18 The most frequently involved mechanism consists of increased INalate.7,18,23 However, there are other mechanisms such as increased INa peak, decreased inactivation rate or increased window current (Figure 3).18,22
Rarely, the mutations may simultaneously cause reduced INapeak and increased INalate, occurring with loss and gain of function, respectively.7,23
Mutations in β subunitsMutations in the β1 subunits were identified in patients with BrS, AF and cardiac conduction disease (CCD) (Table 3). The mechanism involved in these phenotypes is believed to occur with a decrease in INa density (loss of function). However, given the limited number of patients with these mutations, the mechanism involved or the genotype-phenotype relationship cannot be completely determined.20,23,25
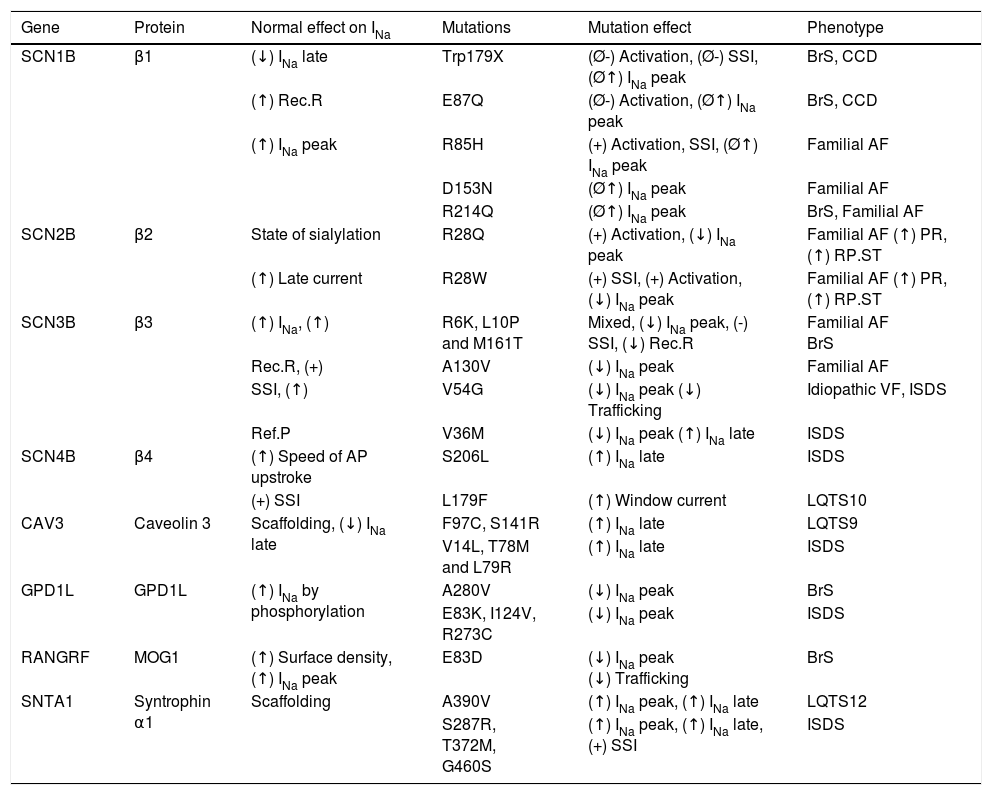
Mutations in proteins of the sodium channel macromolecular complex.
| Gene | Protein | Normal effect on INa | Mutations | Mutation effect | Phenotype |
|---|---|---|---|---|---|
| SCN1B | β1 | (↓) INa late | Trp179X | (Ø-) Activation, (Ø-) SSI, (Ø↑) INa peak | BrS, CCD |
| (↑) Rec.R | E87Q | (Ø-) Activation, (Ø↑) INa peak | BrS, CCD | ||
| (↑) INa peak | R85H | (+) Activation, SSI, (Ø↑) INa peak | Familial AF | ||
| D153N | (Ø↑) INa peak | Familial AF | |||
| R214Q | (Ø↑) INa peak | BrS, Familial AF | |||
| SCN2B | β2 | State of sialylation | R28Q | (+) Activation, (↓) INa peak | Familial AF (↑) PR, (↑) RP.ST |
| (↑) Late current | R28W | (+) SSI, (+) Activation, (↓) INa peak | Familial AF (↑) PR, (↑) RP.ST | ||
| SCN3B | β3 | (↑) INa, (↑) | R6K, L10P and M161T | Mixed, (↓) INa peak, (-) SSI, (↓) Rec.R | Familial AF BrS |
| Rec.R, (+) | A130V | (↓) INa peak | Familial AF | ||
| SSI, (↑) | V54G | (↓) INa peak (↓) Trafficking | Idiopathic VF, ISDS | ||
| Ref.P | V36M | (↓) INa peak (↑) INa late | ISDS | ||
| SCN4B | β4 | (↑) Speed of AP upstroke | S206L | (↑) INa late | ISDS |
| (+) SSI | L179F | (↑) Window current | LQTS10 | ||
| CAV3 | Caveolin 3 | Scaffolding, (↓) INa late | F97C, S141R | (↑) INa late | LQTS9 |
| V14L, T78M and L79R | (↑) INa late | ISDS | |||
| GPD1L | GPD1L | (↑) INa by phosphorylation | A280V | (↓) INa peak | BrS |
| E83K, I124V, R273C | (↓) INa peak | ISDS | |||
| RANGRF | MOG1 | (↑) Surface density, (↑) INa peak | E83D | (↓) INa peak (↓) Trafficking | BrS |
| SNTA1 | Syntrophin α1 | Scaffolding | A390V | (↑) INa peak, (↑) INa late | LQTS12 |
| S287R, T372M, G460S | (↑) INa peak, (↑) INa late, (+) SSI | ISDS |
Ø: failure; (↑): increase; (↓): decrease; (+): depolarizing shift; (-): hyperpolarizing shift; AF: atrial fibrillation; BrS: Brugada syndrome; CCD: cardiac conduction disease; ISDS: infant sudden-death syndrome; LQTS: long QT syndrome; Rec.R: recovery rate; Ref.P: refractory period; RP.ST: ST segment in right precordial leads; SSI: steady state inactivation; VF: ventricular fibrillation.
Extracted and adapted from Adsit et al. (2013).23
The prevalence of potentially pathogenic variants of the genes for the β subunits is similar to that of other minor genes involved in BrS.29,30 Indeed, even though in recent years the knowledge of the underlying mechanisms of BrS focuses mainly on the SCN5A gene, the screening of the four β subunits may lead to a potential increase in the genetic diagnosis of the syndrome, up to approximately 5.4%.30
Mutations in the β1 and β2 subunits are associated with AF, and the mechanism consists of alterations in gating and decreased INa.6,31 In 2011, Olesen et al.32 described mutations associated with AF, also in the β3 subunit. These mutations decrease INa, increasing the susceptibility for AF through one of two mechanisms: conduction delay or decrease in the refractory period (promoting the possibility of reentry circuits).32 Moreover, mutations in SCN3B are also associated with BrS (Table 3).8,23
Mutations in β4 subunit have already been described in LQTS10 and confer gain of function, whose most likely mechanism consists of increased INalate.23,25,30,33
Mutations in proteins associated with sodium channelsThe NaC are part of a macromolecular complex that includes various proteins that participate in cell adhesion, signal transduction pathways and the cytoskeleton (Figure 1).6,7,11 These proteins are directly or indirectly bound to the NaC and have the ability to modulate their expression, traffic and function.6,19,23 Therefore, their dysfunction contributes to the pathophysiology of the cardiac channelopathies.6,22,23,28
In fact, mutations in several of these proteins are associated with LQTS or BrS (Table 3).5,22,34–36 Caveolin-3 (CAV3) is an important protein in membrane traffic and in the positioning of the ion channels in the sarcoplasmatic membrane, which regulates various ion currents in the heart such as INa. Syntrophin α1 (SNTA1) is a cytoskeleton protein that interacts with the NaC (Figure 1). Gain-of-function mutations described in CAV3 are associated with LQTS9, whereas those described for SNTA1 are associated with a phenotype similar to LQTS3.6,15,22,28,37 On the other hand, mutations in ankyrin-B, whose function is to bind membrane proteins to cytoskeleton structures (Figure 1), are associated with LQTS4 and AF, among others.15
Mutations in the GPD1L gene, which encodes the glycerol-3-phosphate dehydrogenase 1-like protein, or in the MOG1 gene, which encodes a molecule that affects protein traffic, have already been described in BrS.6,15,23,25 In addition, mutations in plakophilin-2, a desmosomal protein, may decrease INa and therefore lead to a phenotype similar to BrS.15,38,39
“Cardiac” phenotypes associated with dysfunction in sodium channels and interacting proteinsThe numerous “cardiac” phenotypes associated with mutations in the genes encoding for the NaC and the proteins that make up its macromolecular complex are described in Table 4. The most prevalent cardiac channelopathies are LQTS (1:2500) and BrS (1:3300 to 1:10000), which are partially associated with NaC dysfunction.12 Therefore, we will now cover only these two entities.
“Cardiac” phenotypes associated with dysfunction of sodium channels and related proteins.
| Gene | Protein | Abnormalities in INa | “Cardiac” phenotype |
|---|---|---|---|
| Sodium channel | |||
| SCN5A | Nav1.5 | (↓) INa by different mechanisms | Type 1 BrS |
| (↑) INa late | Type 3 LQTS | ||
| (↓) INa by different mechanisms | CCD | ||
| (↓) INa by different mechanisms | Lev-Lenégre disease | ||
| (↓) INa by different mechanisms | Congenital AV block | ||
| (↓) INa | SND | ||
| (↓) INa | Atrial standstill | ||
| Different and mismatched molecular phenotypes | AF | ||
| Different and mismatched molecular phenotypes | DCM | ||
| (↑) INa late/(↓) INa | ISDS | ||
| Combination of molecular phenotypes present in other clinical entities | Overlap Syndrome | ||
| Proteins of sodium channel macromolecular complex | |||
| SCN1B | Subunit β1 | (↓) INa peak | Type 5 BrS |
| (↓) INa peak | CCD | ||
| (↓) INa peak | AF | ||
| SCN2B | Subunit β2 | (↓) INa peak | AF |
| SCN3B | Subunit β3 | (↓) INa peak | Type 7 BrS |
| (↓) INa peak | AF | ||
| (↓) INa peak | CCD | ||
| (↓) INa peak/(↑) INa late | ISDS | ||
| (↓) INa peak | Idiopathic VF | ||
| SCN4B | Subunit β4 | (↑) INa late | Type 10 LQTS |
| (↑) INa late | ISDS | ||
| SNTA | Syntrophin α1 | (↑) INa late/(↑) INapeak | Type 12 LQTS |
| (↑) INa late/(↑) INapeak | ISDS | ||
| RANGRF | MOG1 | (↓) INa peak | Type 8 BrS |
| CAV3 | Caveolin-3 | (↑) INa late | Type 9 LQTS |
| (↑) INa late | ISDS | ||
| GPD1L | Glycerol-3-phosphate dehydrogenase 1-like | (↓) INa peak | Type 2 BrS |
| (↓) INa peak | ISDS | ||
| PTPH1 | Tyrosine phosphatase H1 | – | |
| NEDD4L | Nedd4-2/Nedd4-like | – | |
| CALM | Calmodulin | – | |
| CAMK2D | Calcium/Calmodulin-dependent protein kinase II delta | – | |
| SAP97 | SAP97 | – | |
| YWHAH | 14-3-3-a | – | |
| FGF13 | FGF13 | – | |
| ANK3 | Ankyrin-G | – | |
| ACTN2 | Actinin α2 | – | |
| PKP2 | Plakophilin-2 | Arrhythmogenic cardiomyopathy | |
| DSG2 | Desmoglein-2 | Arrhythmogenic cardiomyopathy | |
| TCAP | Telethonin | – | |
| ZASP | Z band | – | |
AF: atrial fibrillation; AV: atrioventricular; BrS: Brugada syndrome; CCD: cardiac conduction disease; DCM: dilated cardiomyopathy; ISDS: infant sudden death syndrome; LQTS: long QT syndrome; SND: sinus node disease.
Extracted and adapted from Wilde and Brugada (2011),5 Remme (2013),6 Abriel (2010)15 and Adsit et al. (2013).23
BrS was first described in 1992 as a syndrome characterized by a typical electrocardiographic pattern, absence of structural heart anomalies and family history of SD. Since then, progress has been made in understanding its pathophysiology and in identifying its genetic basis.1,40,41
BrS is a rare hereditary syndrome with an estimated prevalence of 1/3300 to 1/10000, and ethnic and geographic differences have already been described.12,14,42 It affects relatively young adults (<40 years old), more frequently males, with a family history of SD in 20-50% of the cases.21,43,44 Moreover, it is estimated that BrS is responsible for at least 4% of all SD cases and for at least 20% of all SD cases in individuals without structural heart abnormalities.8,45
The absence of structural heart anomalies was classically a characteristic of BrS.41 However, mild structural anomalies in the right and left ventricles have been described in various studies.46
Most individuals are asymptomatic at the time of diagnosis, which is made following a routine ECG in approximately 58% of cases or as a result of family screening in approximately 37% of cases.44 However, SCD may be the first sign of the disease since these individuals are at increased risk for developing tachyarrhythmias, namely PVT and VF.22,47
It is estimated that the rate of arrhythmia events per year in symptomatic individuals is about 0.5%, occurring more frequently at rest and while sleeping, but also in the presence of fever or after a heavy meal.1,44,48 In fact, fever is one of the factors that may cause or exacerbate the electrocardiographic pattern of BrS, triggering potentially fatal arrhythmias in about 27% of cases.21,49
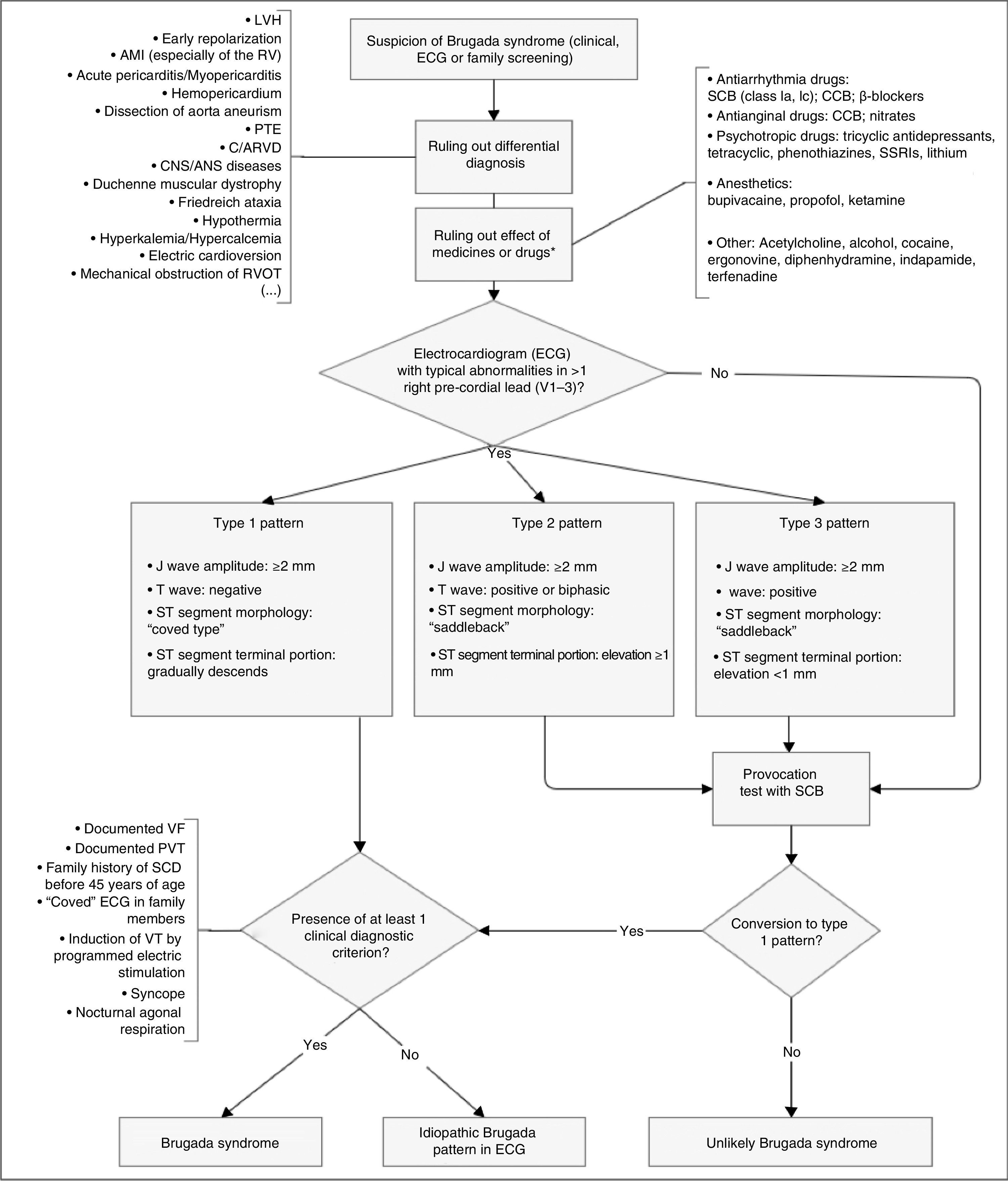
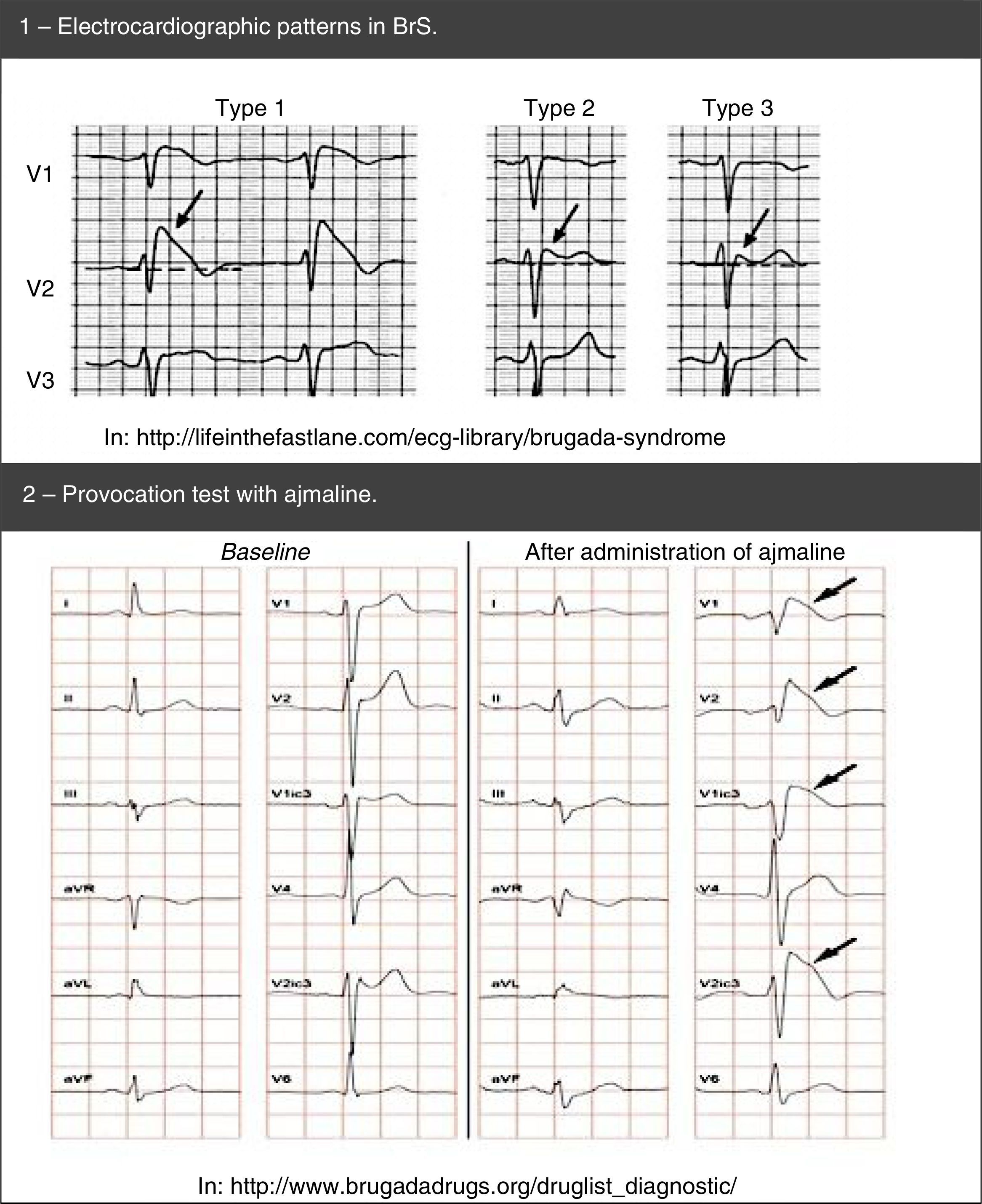
Diagnosis is made using clinical criteria, presence of a typical pattern of electrocardiographic abnormalities and exclusion of other etiologies that may mimic BrS, namely because they trigger ST-segment elevation (Figure 4).45,50,51 Provocation tests with NaC-blocking agents (Table 5) may be performed to provoke the electrocardiographic abnormalities seen in BrS (Figure 5), which enables diagnosing those individuals with transient electrocardiographic pattern.14,41,45,52 In these cases, Holter may also be used and, through prolonged monitoring, it may enable diagnosing intermittent abnormalities.1
 BrS are: prolongation of the PR interval and right branch block. A definitive diagnosis is made in presence of type 1 ST-segment elevation in at least one V1-V3 lead and when one of the clinical criteria presented in the figure is met. AMI: acute myocardial infarction; ANS: autonomic nervous system; C/ARVD: cardiomyopathy/arrhythmogenic right ventricular dysplasia; CCB: calcium channel blockers; CNS: central nervous system;
BrS are: prolongation of the PR interval and right branch block. A definitive diagnosis is made in presence of type 1 ST-segment elevation in at least one V1-V3 lead and when one of the clinical criteria presented in the figure is met. AMI: acute myocardial infarction; ANS: autonomic nervous system; C/ARVD: cardiomyopathy/arrhythmogenic right ventricular dysplasia; CCB: calcium channel blockers; CNS: central nervous system; Diagnostic algorithm for Brugada syndrome. There are three patterns of electrocardiographic abnormalities in the right precordial leads (V1-V3). Type 1 is considered to be diagnostic, unlike types 2 and 3 (in presence of which provocation tests with SCB must be performed). Other electrocardiographic abnormalities that may be present in BrS are: prolongation of the PR interval and right branch block. A definitive diagnosis is made in presence of type 1 ST-segment elevation in at least one V1-V3 lead and when one of the clinical criteria presented in the figure is met. AMI: acute myocardial infarction; ANS: autonomic nervous system; C/ARVD: cardiomyopathy/arrhythmogenic right ventricular dysplasia; CCB: calcium channel blockers; CNS: central nervous system; ECG: electrocardiogram; LVH: left ventricular hypertrophy; PTE: pulmonary thromboembolism; RV: right ventricle; RVOT: right ventricular outflow tract; SCB: sodium channel blockers; SSRIs: selective serotonin reuptake inhibitors; VF: ventricular fibrillation; VT: ventricular tachycardia; β-blockers: beta blockers. *May unmask genetic susceptibility to BrS.
Extracted and adapted from Berne and Brugada (2012).51
Drugs used in provocation tests to “unmask” Brugada syndrome.
| Drug | Dose and duration | Route of administration |
|---|---|---|
| Ajmaline | 1 mg/kg for 5 minutes | IV |
| Flecainide | 2 mg/kg for 10 minutes | IV |
| 400 mg | PO | |
| Pilsicainide | 1 mg/kg for 10 minutes | IV |
| Procainamide | 10 mg/kg for 10 minutes | IV |
IV: intravenous; PO: per os.
Extracted and adapted from Antzelevitch et al. (2005).45
 BrS: spontaneous (1) and after provocation test with ajmaline (2).' title='Electrocardiographic patterns in
BrS: spontaneous (1) and after provocation test with ajmaline (2).' title='Electrocardiographic patterns in Genetic tests (broad or specific for the SCN5A gene) may also be useful for diagnosis in any case where there is strong clinical suspicion of BrS according to the family and medical history and the ECG.53 It should be noted that after identifying a pathogenic mutation in a BrS case index, specific genetic screening of family members is indicated.53,54
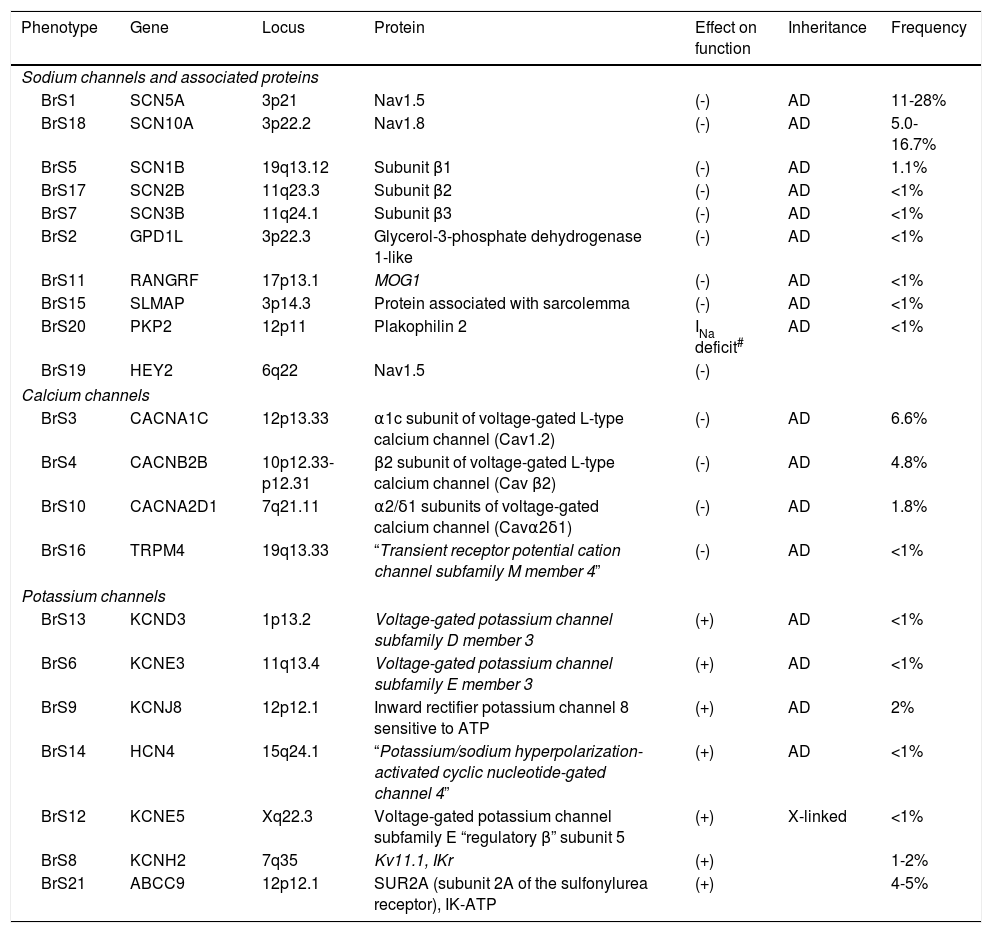
BrS presents high genetic complexity and there are various genes that may be mutated in this syndrome, although only a few are associated with abnormalities in INa (Table 6).43,54,55 As of today, more than 300 mutations reducing INa amplitude through different mechanisms have been described.16,43,54,56 The mutations occur more frequently in the SCN5A gene, and usually occur in transmembrane segments S1-S4 and in the segments involved in pore formation (S5-S6).16,55 However, only a few (approximately 10-30%) of the total number of individuals diagnosed with BrS are positive for a mutation in this gene.21,43,56–58 Other genes (Table 6) are involved in fewer than 5% of the cases.55
Mutated genes in Brugada syndrome.
| Phenotype | Gene | Locus | Protein | Effect on function | Inheritance | Frequency |
|---|---|---|---|---|---|---|
| Sodium channels and associated proteins | ||||||
| BrS1 | SCN5A | 3p21 | Nav1.5 | (-) | AD | 11-28% |
| BrS18 | SCN10A | 3p22.2 | Nav1.8 | (-) | AD | 5.0-16.7% |
| BrS5 | SCN1B | 19q13.12 | Subunit β1 | (-) | AD | 1.1% |
| BrS17 | SCN2B | 11q23.3 | Subunit β2 | (-) | AD | <1% |
| BrS7 | SCN3B | 11q24.1 | Subunit β3 | (-) | AD | <1% |
| BrS2 | GPD1L | 3p22.3 | Glycerol-3-phosphate dehydrogenase 1-like | (-) | AD | <1% |
| BrS11 | RANGRF | 17p13.1 | MOG1 | (-) | AD | <1% |
| BrS15 | SLMAP | 3p14.3 | Protein associated with sarcolemma | (-) | AD | <1% |
| BrS20 | PKP2 | 12p11 | Plakophilin 2 | INa deficit# | AD | <1% |
| BrS19 | HEY2 | 6q22 | Nav1.5 | (-) | ||
| Calcium channels | ||||||
| BrS3 | CACNA1C | 12p13.33 | α1c subunit of voltage-gated L-type calcium channel (Cav1.2) | (-) | AD | 6.6% |
| BrS4 | CACNB2B | 10p12.33-p12.31 | β2 subunit of voltage-gated L-type calcium channel (Cav β2) | (-) | AD | 4.8% |
| BrS10 | CACNA2D1 | 7q21.11 | α2/δ1 subunits of voltage-gated calcium channel (Cavα2δ1) | (-) | AD | 1.8% |
| BrS16 | TRPM4 | 19q13.33 | “Transient receptor potential cation channel subfamily M member 4” | (-) | AD | <1% |
| Potassium channels | ||||||
| BrS13 | KCND3 | 1p13.2 | Voltage-gated potassium channel subfamily D member 3 | (+) | AD | <1% |
| BrS6 | KCNE3 | 11q13.4 | Voltage-gated potassium channel subfamily E member 3 | (+) | AD | <1% |
| BrS9 | KCNJ8 | 12p12.1 | Inward rectifier potassium channel 8 sensitive to ATP | (+) | AD | 2% |
| BrS14 | HCN4 | 15q24.1 | “Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 4” | (+) | AD | <1% |
| BrS12 | KCNE5 | Xq22.3 | Voltage-gated potassium channel subfamily E “regulatory β” subunit 5 | (+) | X-linked | <1% |
| BrS8 | KCNH2 | 7q35 | Kv11.1, IKr | (+) | 1-2% | |
| BrS21 | ABCC9 | 12p12.1 | SUR2A (subunit 2A of the sulfonylurea receptor), IK-ATP | (+) | 4-5% | |
AD: autosomal dominant.
In fact, only 30-35% of the individuals with a clinical diagnosis have a genetic diagnosis as well (positive genotype).8 Therefore, the majority of the individuals affected (approximately 65%) remains genetically undetermined (negative genotype) and for this reason identifying new susceptibility genes for BrS is necessary.16,56,59
Recently, the SCN10A gene was identified as a susceptibility gene for BrS, although its real prevalence has yet to be determined.56,60 The expression level and function of the Nav1.8 NaC in the heart are still controversial. However, a study published in 2014 shows that the variants of this gene influence the duration of the PR and QRS interval, heart rate (HR) and also the risk of arrhythmias.56
A few recessive forms with homozygous or compound heterozygous mutations have been described, but most of the known pathogenic mutations in the SCN5A gene present an autosomal dominant transmission pattern with variable, and frequently incomplete, penetrance.18,43,45
The mechanism most frequently implicated is decreased INapeak due to mutations in the SCN5A gene (loss of function) and consequent slowing of cardiac conduction (Figure 3).7,18,60,61 Nevertheless, there are many hypotheses for the pathophysiological mechanisms of BrS that involve depolarization and repolarization abnormalities; however, the latter are not covered in this article.40,51,54
The genotype of individuals with BrS does not currently carry relevant implications for prognosis or treatment (Table 7).9 Nonetheless, its influence in the risk of arrhythmia and in prognosis is still under debate.54 In reality, genetic data may constitute a complementary tool for risk stratification.43,61 Nonsense mutations, which result in truncated proteins, have been associated with a poorer prognosis compared with other types of mutations with less marked repercussions in NaC function.40,43,59,61 A retrospective study published in 2009 shows that the phenotype is more severe in individuals with mutations associated with more significant INa reductions compared with individuals with mutations associated with lower reductions.61 The same is seen when the mutation is located in a transmembrane region of the NaC.61 Another study published in 2013 shows that different mutations in the SCN5A gene have a different impact on INa, emphasizing the role of mutation characterization in the risk assessment for nonaffected family members.62 However, it is still not clear to what extent different mutations confer a risk for arrhythmia events or SCD, thus the risk is currently stratified only with clinical parameters.53,54
Recommendations for the treatment of Brugada syndrome.
| General measures for lifestyle alterations | |
|---|---|
| Avoid drugs that may induce or worsen ST segment elevation in RPCld | (Class I) |
| Avoid excessive alcohol consumption | (Class I) |
| In presence of fever, promptly medicate with antipyretic drug | (Class Ia) |
| Risk stratification and specific treatment | |||
|---|---|---|---|
| Symptomatic individualsa | Asymptomatic individuals | ||
| “aborted” SCD | ICD (Class I) | Spontaneous type 1 electrocardiogram (ECG) pattern | Quinidine (Class IIb) |
| Documented spontaneous VT, with or without syncope | ICD (Class I) | Spontaneous type 1 ECG pattern + VT/VF induced by EPS | ICD (Class IIb) |
| Syncope + Spontaneous type 1 ECG pattern | ICD (Class IIa) | ||
| Electrical/arrhythmic stormb | Isoprenalinec (Class IIa) | Type 1 ECG pattern induced by drugs and family history of SCD | ICD (Class III) |
| Quinidined (Class IIa) | |||
| Individuals who are eligible for ICD but present a contraindication or refuse ICD and/or present with a history of supraventricular arrhythmias that require treatment | Quinidine (Class IIa) | ||
| Individuals diagnosed with BrS and history of electrical/arrhythmic storms or (appropriate) repetition shocks due to ICD | Catheter ablation – RF (Class IIb) | ||
BrS: Brugada syndrome; ICD: implantable cardioverter defibrillator; RF: radiofrequency; RPCld: right precordial leads; SCD: sudden cardiac death; VF: ventricular fibrillation; VT: ventricular tachycardia.
The only treatment available proven capable of preventing SCD in patients with BrS was the implementation of an implantable cardioverter defibrillator (ICD).1,9,44,46 This procedure, however, results in a considerable risk of complications, which occur in approximately 9% of patients/year and, although rarely life-threatening, they are psychologically harmful.9,54 Therefore, a careful assessment of risks (namely the risk of arrhythmia) and benefits is a key process in this decision.44,54
A 2003 study that included 547 individuals diagnosed with BrS, showing a diagnostic electrocardiographic pattern but no prior “aborted” SCD, was conducted with the aim of assessing the prognostic value of clinical, electrocardiographic and electrophysiological variables. The authors observed that the group with lower risk (incidence of events: 0.5%) is characterized by absence of syncope episodes, an electrocardiographic pattern only triggered by antiarrhythmics and absence of arrhythmia during programmed ventricular stimulation (PVS). However, the group with higher risk (incidence of events: 27.2%) is characterized by prior history of syncope episodes, spontaneously abnormal ECG and presence of arrhythmias induced by PVS. Moreover, individuals with inducibility of arrhythmias in PVS have a six-fold higher risk of SCD or VF during the subsequent two years than those who do not have it.63
Although some are controversial, there are many risk factors for arrhythmia events, and among them, symptoms are one of the most important.42,46,54,64 In fact, individuals diagnosed after an “aborted” SCD are at the highest risk, and in approximately 60% of these cases there is a new event 10 years after the diagnosis.54 Individuals with syncope episodes have a rate of arrhythmia events of 1.9%/year, and the simultaneous presence of a type 1 electrocardiographic pattern is associated with a poor prognosis.46,54 Additionally there are other electrocardiographic parameters that are associated with a poorer prognosis, for example, presence of QRS-interval fragmentation in the ECG, identified in 30-40% of the patients.54
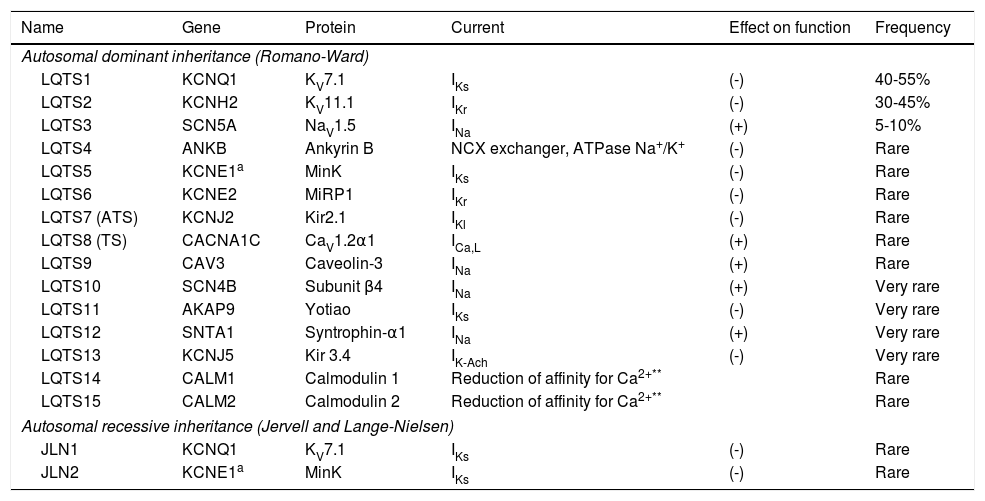
Long QT syndromeCongenital LQTS is an arrhythmia syndrome with a genetic/hereditary etiology and incomplete penetrance, whose prevalence in Caucasians is approximately 1:2500, a value much higher than what was previously expected.12,13,65 It represents a heterogeneous group of diseases and, classically, it is divided into two variants: Romano-Ward syndrome and Jervell and Lange-Nielsen syndrome (Table 8).1,43,65
Subtypes of congenital LQTS.
| Name | Gene | Protein | Current | Effect on function | Frequency |
|---|---|---|---|---|---|
| Autosomal dominant inheritance (Romano-Ward) | |||||
| LQTS1 | KCNQ1 | KV7.1 | IKs | (-) | 40-55% |
| LQTS2 | KCNH2 | KV11.1 | IKr | (-) | 30-45% |
| LQTS3 | SCN5A | NaV1.5 | INa | (+) | 5-10% |
| LQTS4 | ANKB | Ankyrin B | NCX exchanger, ATPase Na+/K+ | (-) | Rare |
| LQTS5 | KCNE1a | MinK | IKs | (-) | Rare |
| LQTS6 | KCNE2 | MiRP1 | IKr | (-) | Rare |
| LQTS7 (ATS) | KCNJ2 | Kir2.1 | IKl | (-) | Rare |
| LQTS8 (TS) | CACNA1C | CaV1.2α1 | ICa,L | (+) | Rare |
| LQTS9 | CAV3 | Caveolin-3 | INa | (+) | Rare |
| LQTS10 | SCN4B | Subunit β4 | INa | (+) | Very rare |
| LQTS11 | AKAP9 | Yotiao | IKs | (-) | Very rare |
| LQTS12 | SNTA1 | Syntrophin-α1 | INa | (+) | Very rare |
| LQTS13 | KCNJ5 | Kir 3.4 | IK-Ach | (-) | Very rare |
| LQTS14 | CALM1 | Calmodulin 1 | Reduction of affinity for Ca2+** | Rare | |
| LQTS15 | CALM2 | Calmodulin 2 | Reduction of affinity for Ca2+** | Rare | |
| Autosomal recessive inheritance (Jervell and Lange-Nielsen) | |||||
| JLN1 | KCNQ1 | KV7.1 | IKs | (-) | Rare |
| JLN2 | KCNE1a | MinK | IKs | (-) | Rare |
(-): loss of function; (+): gain of function; ATS: Andersen-Tawil syndrome; ICa,L: Ca2+ currents through voltage-gated type-L calcium channels; IK-Ach: K+ current regulated by acetylcholine receptors; IKl: K+ entry current, rectifying; IKr: rapid component (internal rectification – K+ channels are open when potential is negative and closed when potential is less negative or positive) of the K+ “delayed rectifier” current (IKr); IKs: slow component of the K+ “delayed rectifier” current (IKr); INa: voltage-gated Na+ current; NCX: Na+/Ca2+ exchanger; TS: Timothy syndrome.
Mutations in the KCNE1 gene may cause Romano-Ward syndrome (autosomal dominant; LQTS5) or, if homozygous or compound heterozygous, Jervell and Lange-Nielsen syndrome (autosomal recessive).
Calmodulin dysfunction may alter the inactivation of Ca2+-gated L-type Ca2+ channels (increasing the depolarizing current during phase 2 of the action potential), but some calmodulin mutations may also be associated with an abnormality in the regulation of sodium channels.
Extracted and adapted from Nakano and Shimizu (2016),34 **Makita et al. (2014)36 and *Mizusawa (2014).69
In 1995 and 1996, the three main genes conferring susceptibility to LQTS were identified: KCNQ1, KCNH2 and SCN5A.66–68 These genes constitute about 75% of the clinically defined LQTS and the remainder collectively represents only 5% of these cases.43,69 It should be noted that LQTS is associated with abnormalities in sodium currents only for types 3, 9, 10 and 12.22,69
LQTS is characterized by a delay in ventricular repolarization, which translates echocardiographically as QT interval prolongation (Figure 6).22,65 The duration of the QT interval depends on NaC inactivation, the alteration of which may trigger arrhythmias.26
Mutations in the SCN5A gene associated with LQTS3 (gain of function) usually affect NaC inactivation, which is slower, unstable or incomplete.6,18,22,26 Consequently, there is an increase in INalate with prolongation of membrane depolarization and delay in repolarization.18,26,65,70 Other mechanisms potentially involved are: increased window current, slower inactivation and increased INapeak (Figure 3).6,18,22
The first mutation associated with LQTS3 is found in the loop between domains III and IV, corresponding to the inactivation gate.26 Since then, multiple mutations causing abnormalities in inactivation have been identified and functionally characterized. They may be located at different sites in the NaC structure, namely in the C terminus, to which a relevant function in this process has been attributed.22,26
Congenital LQTS occurs mainly in young, healthy individuals without concomitant structural heart abnormalities and is associated with an increased risk of syncope and potentially fatal heart arrhythmias such as Torsade de Pointes (TdP), which degenerates into VF and causes cardiac arrest.6,18,66 In LQTS3 (unlike LQTS1 and LQTS2 – Figure 6), arrhythmias usually occur at rest, particularly while sleeping (low HR).6,22,43,70 It should be noted that INalate is higher with slower stimulation frequencies, suggesting that the intensity of this current may be a strong factor in determining the occurrence of arrhythmias.22
The first cardiac event (more frequently syncope) usually occurs in adolescents (16±10 years old in LQTS3) and earlier among males.9,71 However, in approximately 5-10% of cases, SCD is the initial event of the disease and, actually, LQTS is one of the main causes of SCD with negative autopsy.10,72
Diagnosis is mainly based on medical history and ECG (Figures 6 and 7).65,70 In ECGs, the QT interval is the most relevant parameter (Table 9), and is measured from the beginning of the QRS complex to the end of the T wave in the DII and V5 or V6 leads.65,73 The longest value is used, generally corrected for HR (QTc) with the Bazett formula (despite its limitations for particularly rapid or slow HR).1,46,65,73
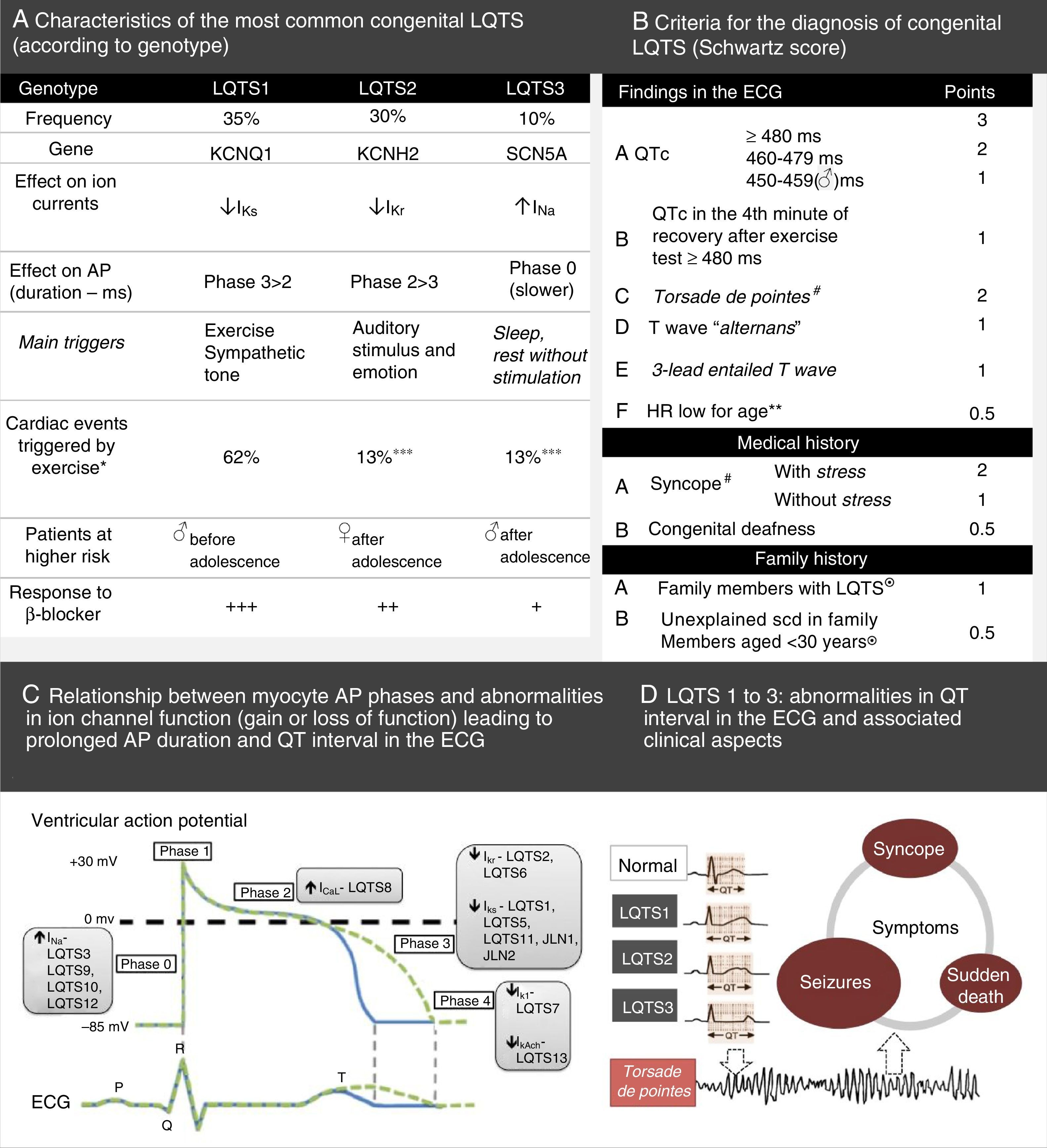
 LQTS based on the findings in the electrocardiogram, medical history (symptoms) and family history.
LQTS based on the findings in the electrocardiogram, medical history (symptoms) and family history. Long QT syndrome – main characteristics and diagnostic criteria. (B) Scoring system used in the diagnosis of LQTS based on the findings in the electrocardiogram, medical history (symptoms) and family history. QTc is calculated using the Bazett formula. Scoring: ≤1 – low likelihood of LQTS; 1.5 to 3 – intermediate likelihood of LQTS; ≥3.5 – high likelihood of LQTS. LQTS is diagnosed in individuals with a score ≥3.5 in whom there are no secondary causes for QT interval prolongation.
♂: male individuals; ♀: female individuals; HR: heart rate.
#Mutually exclusive. ¿Both cannot be accounted for in the same family.
**HR at rest below the 2nd percentile for age.
***The low risk associated with exercise in LQTS2 and LQTS3 patients is explained by the fact that both have a normal IK current, stimulated by activation of the sympathetic nervous system, which in turn results in shortening of the ventricular repolarization whenever the heart rate increases, thus avoiding the likelihood of ventricular tachyarrhythmias during exercise.
(A) Extracted and adapted from Furst and Aziz (2016)75 and *Schwartz et al. (2001)74; (B) Extracted and adapted from Schwartz et al. (2013)43; (C) and (D) Extracted and adapted from Giudicessi and Ackerman (2013).70
.70")
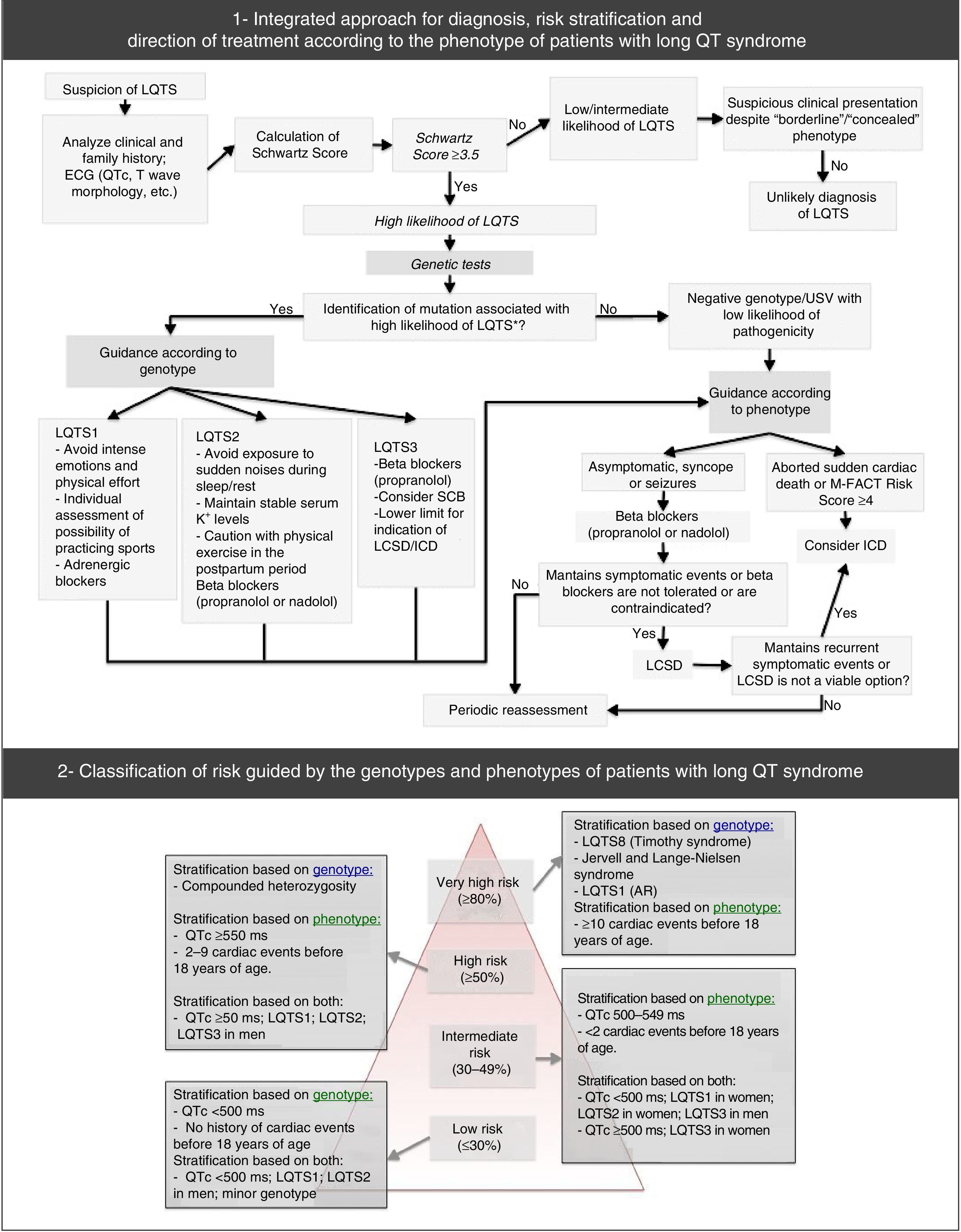
Diagnostic approach, risk stratification and guidance for the treatment of long QT syndrome.
USV: undetermined significance variant. Extracted and adapted from Giudessi and Ackerman (2013).70
Assessment of the QT interval.
| 1 – Method for correction of QT interval (formulas) | |
|---|---|
| Bazett | QT/RR1/2 |
| Fridericia | QT/RR1/3 |
| Framingham | QT + 0.154 (1 - RR) |
| Hodges | QT + 1.75 (HR - 60) |
| 2 – Normal, borderline and prolonged QTc values calculated with the Bazett formula | |||
|---|---|---|---|
| Normal | Borderline | Prolonged | |
| 1-15 years | <440 ms | 440-460 ms | >460 ms |
| Adult (♂) | <430 ms | 430-450 ms | >450 ms |
| Adult (♀) | <450 ms | 450-470 ms | >470 ms |
♂: male; ♀: female; HR: heart rate; RR: RR interval.
Extracted and adapted from Goldenberg et al. (2006).73
Additionally, secondary causes for QT interval prolongation (acquired LQTS) must be ruled out, for example: drugs, myocardial ischemia, cardiomyopathy, hypokalemia, hypomagnesemia and hypothermia, among others.46,65 Once ruled out, the presence of a repetition of a QTc value ≥500 ms (or 480-499 ms if conducted after an unexplained syncope episode) in an electrocardiogram is considered to be diagnostic.43,46 However, LQTS types 1, 2 and 3 may occur with normal QTc in the ECG at rest in 36%, 19% and 10% of cases, respectively.69
A scoring system considering various clinical and electrocardiographic parameters was created for diagnosis and provides the likelihood for LQTS (Figure 6).43,46,70 In addition, Holter monitoring and the ECG obtained during an effort test or after adrenaline infusion may be useful in some particular cases.1,46,53,65,69
Once the diagnosis is made or given the occurrence of unexplained SD in a young individual, first-degree family members must be screened for LQTS.1,65,72 However, LQTS cannot be ruled out in family members with a normal ECG.43 In fact, after identifying a pathogenic mutation in an index case, family members should undergo a genetic test specific for the mutation in question, with the aim of identifying individuals with a normal QT interval.43,53 This is important given the risk of arrhythmias, which is estimated to occur in 10% of asymptomatic carriers.1
Moreover, genetic tests specific for LQTS (broad or specific for the three main genes) are recommended for any patient when there is a strong clinical suspicion of LQTS (based on the medical and family history and the ECG), or for any asymptomatic patient with QT interval prolongation in the absence of other clinical conditions that may prolong this interval.53 In reality, genetic tests play an important role, not only in the diagnosis of LQTS (namely of asymptomatic carriers) and in ruling out disease for first-degree family members, but also in risk stratification, prognosis and treatment (according to the genotype – Figure 7).1,43,69,70,72
Risk stratification takes into account phenotype and genotype and is conducted for all patients with regular clinical assessments (Figure 7).46,65,70 Risk varies according to genotype and, additionally, in the most common genetic types, it is influenced by the specific type and location of the mutations, as well as by the degree of dysfunction that they cause.43,46
Priori et al. (2003)71 followed up 647 individuals with mutations in the genes for LQTS types 1, 2 and 3, for an average period of 28 years. The authors found that 42% of the individuals with LQTS3 developed a first cardiac event (occurrence of syncope, cardiac arrest or SCD) before turning 40 years old and before starting treatment. The incidence of cardiac arrest or SCD in patients with LQTS3 was about 16%, and men presented with symptoms earlier than women. However, given the small study sample, no conclusions could be reached about this finding. Additionally, the authors found that the QTc interval of patients with cardiac events was significantly longer than that of asymptomatic patients (LQTS3 subgroup: 523±55 ms vs. 481±38 ms, p=0.003). They also concluded that only a QTc value higher than 498 ms is associated with a markedly increased likelihood of cardiac events. However, the percentage of individuals in the LQTS3 subgroup with a normal QT interval and carrying a silent mutation was 10%.71
The result of the genetic tests is also important in the treatment and counseling of affected individuals and family members (Figure 7; Table 10).9,43 It should be noted, for example, that LQTS1 involves higher risk during physical activity compared with LQTS2 and LQTS3.43,74,75
Recommendations for the treatment of long QT syndrome.
| General measures for lifestyle alterations | |
|---|---|
| Avoid drugs that prolong the QT interval | (Class I) |
| Identify and correct hydroelectrolytic disorders | (Class I) |
| Risk stratification and specific treatment | |||
|---|---|---|---|
| Symptomatic individuals | Asymptomatic individuals | ||
| Syncope | β-blockers (Class I) | QTc ≥470 ms | β-blockers (Class I) |
| Documented VT/VF | β-blockers (Class I) | QTc ≤470 ms | β-blockers (Class IIa) |
| “aborted” SCD | ICD (Class I) | Not treated with β-blockers* | ICD (Class III) |
| Recurrent syncope episodes during treatment with β-blockers | ICD (Class IIa) | ||
| Individuals with a diagnosis of LQTS who present with events during treatment with β-blockers/ICD | LCSD (Class IIa) | ||
| High-risk individuals with a diagnosis of LQTS who refuse ICD or for whom it is contraindicated and/or when β-blockers are not effective in preventing syncope/arrhythmias, are not tolerated, are contraindicated or are refused | LCSD (Class I) | ||
| Individuals with type 3 LQTS and QTc >500 ms that decrease >40 ms after acute oral test with an SCB | SCB (Class IIa) | ||
ICD: implantable cardioverter defibrillator; LCSD: left cardiac sympathetic denervation; SCB: sodium channel blocker; SCD: sudden cardiac death; VF: ventricular fibrillation; VT: ventricular tachycardia.
Except under special circumstances, the ICD is not indicated in asymptomatic individuals not undergoing treatment with β-blockers.
Adapted from Priori et al. (2013).46
Mexiletine, flecainide or ranolazine constitute “specific” therapeutic options for LQTS3 (Table 10), for which β-blockers may not be so effective, since the adrenergic stress in this type is a trigger with less influence.1,43,70,76 Mexiletine may be used as an add-on to β-blockers.9,43,46,50 However, its effect depends on the type of mutation and may not be beneficial for all individuals with LQTS3.9,12,70
In fact, of the three main types of LQTS, type 3 is the one that involves a higher arrhythmia recurrence rate in individuals receiving treatment with β-blockers (10-15%).70 This justifies the need for individuals with LQTS3 to undergo other invasive procedures, such as left cardiac sympathetic denervation and/or ICD implantation more frequently (Table 10).70
ConclusionsCardiac channelopathies are not common in clinical practice (although they are more common than once thought), but they have a significant impact on quality of life and survival.1,13 The clinical approach constitutes a challenge, owing to their high clinical and genetic heterogeneity.1,76
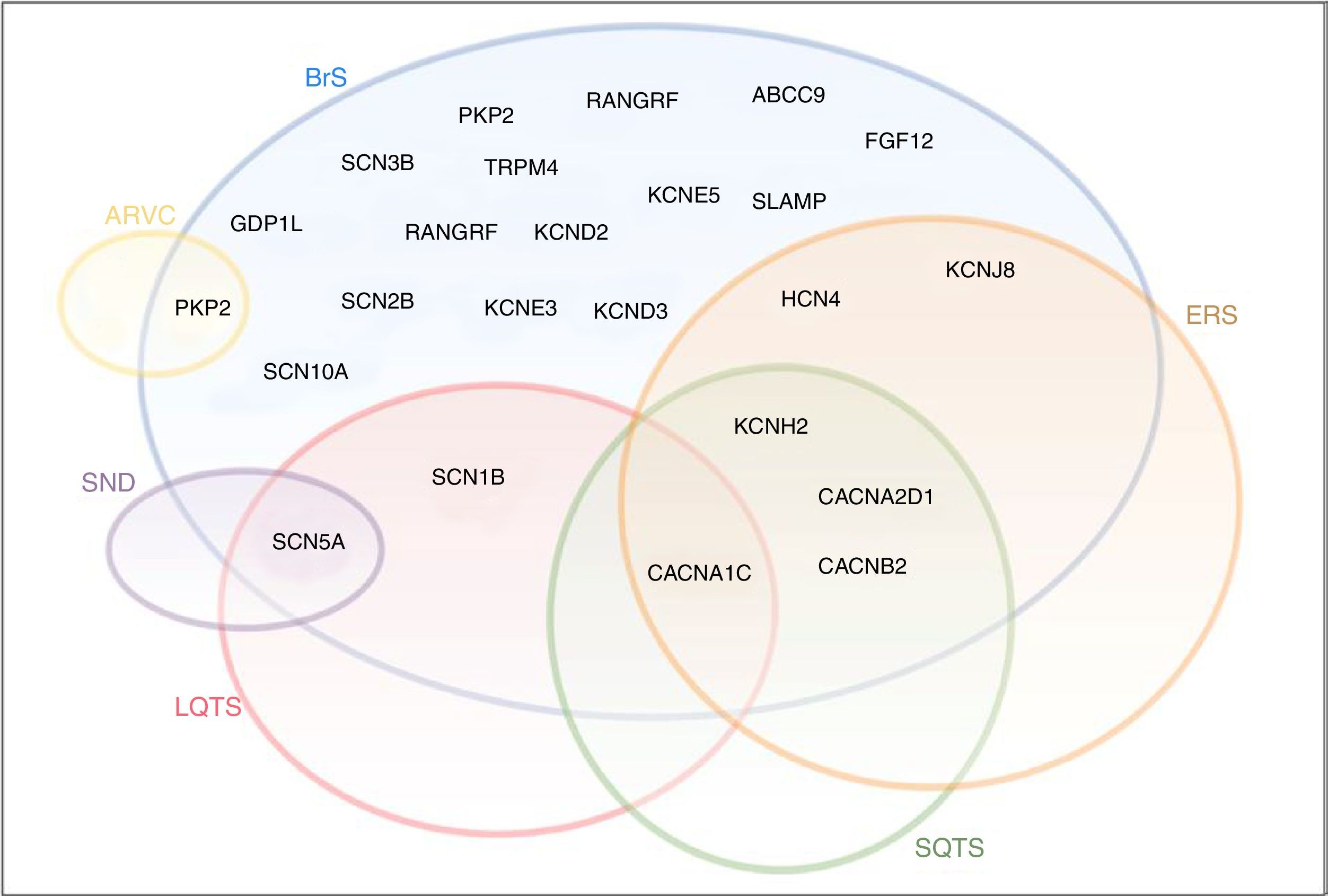
Although they were initially considered to be separate clinical entities with distinct phenotypes, these syndromes may have overlapping clinical and genetic presentations (Figure 8).21 In fact, in addition to strictly loss- or gain-of-function mutations, there is a wide spectrum of mutations associated with various anomalies with different repercussions in NaC function.25 In some cases, a single mutation in the SCN5A gene may result in multiple rhythm disorders, and various phenotypes may thus coexist in the same family.6,25
 BrS,
BrS, Diagram illustrating the overlap between BrS, LQTS, SQTS, SND, ERS and ARVC. The sodium channel macromolecular complex genes are in bold.
ARVC: arrhythmogenic right ventricular cardiomyopathy; ERS: early repolarization syndrome; LQTS: long QT syndrome; SND: sinus node disease; SQTS: short QT syndrome.
Extracted and adapted from Sarquella-Brugada (2016)8 and Fernandez-Falgueras (2017).35
Moreover, some studies have recently reported structural heart anomalies secondary to mutations in this gene (namely DCM), although the underlying mechanism is unknown.6,18,22,77
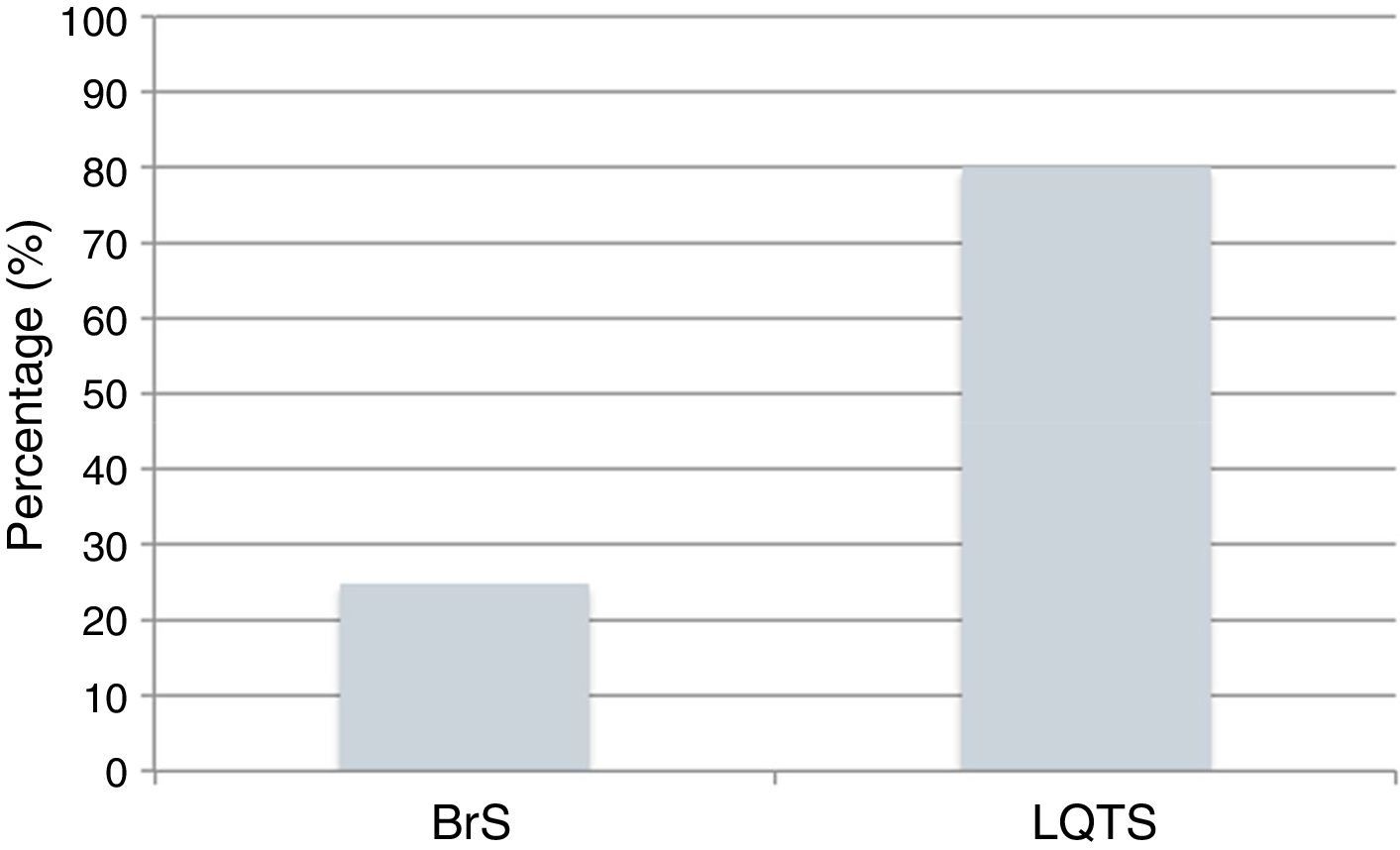
The power of the genetic tests to identify mutations is currently 25% for BrS and 80% for LQTS (Figure 9).43,78 The impact of genetics in clinical approach varies considerably depending on the underlying channelopathies, and is more marked in LQTS, where influence at the level of diagnosis, prognosis and treatment is recognized.9,43,78
 BrS and
BrS and Cardiac channelopathies: positivity of the genetic tests in individuals clinically diagnosed with BrS and LQTS.
Data presented in the charts from Schwartz and Dagradi (2016).78
Great progress has been made in understanding the genotype-phenotype relationship and its implications.43 However, despite increased scientific knowledge in this field, the genotype of a considerable number of affected individuals remains undetermined, some mechanisms still need to be clarified and the treatment options currently available are still limited.12,18,72 A better understanding of molecular principles may contribute not only to increasing knowledge of these aspects, but also to developing new specific treatment approaches for the gene or mutation.12,43
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Fonseca DJNdO, Silva MJLVd. Canalopatias cardíacas: o papel das mutações nos canais de sódio. Rev Port Cardiol. 2018;37:179–199.
