
In adults, unexplained left ventricular hypertrophy is usually due to sarcomeric hypertrophic cardiomyopathy (HCM). Fabry disease (FD) is rare but may mimic sarcomeric HCM, and has an adverse prognosis in the absence of specific treatment. We aimed to assess cardiologists’ awareness of FD based on data from the Portuguese Registry of Hypertrophic Cardiomyopathy.
MethodsA total of 811 index patients, aged 55 ± 16 years, 486 (59.9%) male, were included. Three groups were characterized: A – 128 patients, 74 (57.8%) male, with pathogenic or likely pathogenic mutation(s) in sarcomeric genes; B – 234 patients, 146 (62.4%) male, with negative genetic testing; and C – 449 patients, 266 (59.2%) male, no genetic testing performed. The groups were compared in terms of whether FD was excluded in the registry. Potential red flags for FD were also analyzed and compared between groups.
ResultsPatients in group A were younger and more frequently had familial HCM (A – 53.9% vs. B – 20.1% vs. C – 18.3%; p <0.001). FD was recorded as excluded in 217 (26.8%), similar in all groups; GLA gene testing was performed in only 50/217 patients (A – 48.6%, B – 25.7%, p = 0.019; C – 13.4%, p = 0.036 for B vs. C), mostly in women (p <0.001) in groups B and C. Alpha-galactosidase A (α-Gal A) activity was assessed in 39/217 (18%) patients, with no difference between groups, but more often in men (p = 0.005). Among patients with potential red flags for FD, only 46.7% underwent specific tests (GLA gene testing and/or α-Gal A activity). When GLA genotyping was performed no mutations were identified.
ConclusionsThere is a need to improve cardiologists’ alertness for the identification of FD among the Portuguese HCM population.
Em adultos, hipertrofia ventricular esquerda inexplicada é geralmente devida a miocardiopatia hipertrófica sarcomérica (MH). A doença de Fabry (DF), rara, pode mimetizar MH e tem prognóstico adverso na ausência de tratamento específico. Avaliámos a perceção dos cardiologistas para DF com base no Registo Português de Miocardiopatia Hipertrófica.
MétodosIncluímos 811 doentes-índice, 55 ± 16 anos, 486 (59,9%) homens (H). Caracterizámos três grupos: A-128 doentes, 74 (57,8%) H, com mutação patogénica/provavelmente patogénica em genes sarcoméricos; B-234 doentes, 146 (62,4%) H, com teste genético negativo; C-449 doentes, 266 (59,2%) H, sem teste genético efetuado. Os grupos foram comparados em relação à exclusão de DF, segundo a informação do registo. Sinais potenciais de alerta para DF foram também avaliados e comparados entre os três grupos.
ResultadosOs doentes do grupo A eram mais novos e tinham mais frequentemente MH familiar (A-53,9% versus B-20,1% versus C-18,3%; p<0,001). DF foi dada como excluída em 217 (26,8%) doentes, sem diferença entre grupos; sequenciação do gene GLA foi efetuada apenas em 50/217 doentes [A-48,6%, B-25,7%, p = 0,019; C-13,4%, p (B versus C) = 0,036], predominantemente em mulheres (p < 0,001) nos grupos B e C; atividade enzimática da α-Gal A foi avaliada em 39/217 (18%) doentes, sem diferença entre grupos, mas predominantemente em H (p = 0,005). Dos doentes com sinais potenciais de alerta para DF, apenas 46,7% foram submetidos a testes específicos (GLA e/ou α-Gal A). Quando o gene GLA foi estudado, o resultado foi negativo.
ConclusõesÉ necessário melhorar a perceção dos cardiologistas para a identificação da DF na população portuguesa com MH.
Left ventricular hypertrophy (LVH) detected by imaging techniques in the absence of common physiological or pathological causes is due in about 40-60% of cases to sarcomeric hypertrophic cardiomyopathy (HCM),1 an autosomal dominant trait caused by mutations in cardiac sarcomeric protein genes. The disease has an estimated worldwide prevalence of 1 in 500 individuals on the basis of the echocardiographic phenotype.1 It only affects the heart and is often benign throughout life, although it may also be associated with adverse outcomes, including sudden cardiac death (SCD).1,2 However, in about 5-10% of cases, unexplained LVH can be caused by other non-genetic or rarer genetic disorders that may mimic sarcomeric HCM, for some of which specific treatment is available. This is the case with Fabry disease (FD),3–5 a multisystem lysosomal storage disease caused by a deficiency of the enzyme alpha-galactosidase A (α-Gal A), encoded by the GLA gene on the X chromosome. The disease leads to accumulation of globotriaosylceramide (Gb3) and other glycosphingolipids in lysosomes, resulting in progressive multiorgan damage, with severe complications due to cardiac, renal or cerebrovascular lesions, and ultimately in decreased life expectancy.6–8 Although the classic form of FD affects multiple organs and occurs early in men (hemizygotes), some forms are found particularly (though not exclusively) in women (heterozygotes), manifesting as milder disease, with a late-onset phenotype that is often confined to a single organ such as the heart, kidney, or brain.9–11 The frequent observation of exclusively cardiac involvement in FD suggests that the heart is the most susceptible organ to α-Gal A deficiency.12–14 Cardiac involvement is one of the main determinants of prognosis,6,9,15,16 and includes LVH (the most common manifestation),17 arrhythmias, small-vessel coronary disease and heart failure. Electrocardiographic (ECG) abnormalities are frequent, and brady- or tachyarrhythmias are a significant cause of morbidity and mortality, including SCD.18,19
The diagnosis of FD is difficult and requires a high level of clinical suspicion. Many of the typical extracardiac features may not be evident in non-classical forms of the disease, and in some patients cardiac involvement may predominate, commonly mimicking sarcomeric HCM.20–22 Unlike the latter, LVH is typically concentric in FD,23,24 but it may also manifest as asymmetric septal hypertrophy or even predominantly with involvement of other wall segments.11,13,21,25–27 Left ventricular outflow obstruction (a common feature in sarcomeric HCM)1 may also exist in FD,21 hindering the differential diagnosis between the two conditions.
The accepted prevalence of FD in patients presenting with unexplained LVH is 0.5%-1%, although higher in some series,20,26,28,29 and its exclusion should be considered in this context.11,28,30 When initiated early, enzyme replacement therapy with agalsidase alpha or agalsidase beta can halt the progression of FD and modify its prognosis.3,4,31,32
In men, the first diagnostic test should be measurement of α-Gal A (in plasma, white blood cells or dried blood spots),33 which is generally decreased or absent, followed by GLA gene sequencing. In women, interpretation of enzyme activity results is difficult – only about 40% of female patients have a low enzyme level,34 and sequencing of the GLA gene is necessary for diagnosis.35
In the Portuguese Registry of Hypertrophic Cardiomyopathy (PRo-HCM),36 the possibility of FD was considered in the differential diagnosis of LVH. The aim of the present study was to analyze the registry data in order to assess the awareness of cardiologists regarding the need to exclude this phenocopy in real-world scenarios.
MethodsPRo-HCM was a national multicenter registry designed to collect information on the current approach to sarcomeric HCM in Portugal and to facilitate future improvements regarding diagnosis and therapeutic management of the condition.36 The registry was designed and implemented by the Working Group on Myocardial and Pericardial Diseases of the Portuguese Society of Cardiology (SPC), and centralized and managed at the SPC's National Center for Data Collection in Cardiology (CNCDC). It was an observational, multicenter, voluntary, non-mandatory study, with a two-year enrollment period (from 25 April 2013 to 25 April 2015), largely retrospective but also including a prospective update. Participating centers (n = 29) were asked to include all patients with a diagnosis of HCM followed at the center at that time or in the past (with no retrospective time limit), including those already deceased at the time of enrollment. Included individuals were aged over 18 years at inclusion and had a diagnosis of HCM with LVH phenotype by imaging methods (unexplained LVH with maximum left ventricular wall thickness of ≥15 mm in one or more myocardial segments in index patients).1,37 Criteria for non-inclusion in the registry were the presence of secondary LVH (at least stage 2 hypertension,38 moderate or severe aortic valve stenosis,39 or a previously diagnosed cardiac or systemic disease associated with LVH). However, the electronic case report form included some information about the possible exclusion of FD. This was a non-mandatory open-ended yes/no question, at the discretion of the investigator and not requiring any specific clinical or imaging characterization. However, if the answer was yes, a reference to the specific tests carried out for that purpose (measurement of α-Gal A activity and/or GLA gene sequencing) was requested.
The registry complied with the principles of the Declaration of Helsinki (October 2000) and written informed consent was obtained from all living patients before inclusion.
For the purpose of the present study, only index patients included in the registry were considered. This population was first identified and characterized according to whether sarcomeric genetic testing had been performed and the results thereof. They were then divided into three groups: group A – patients with positive sarcomeric genetic testing, in whom pathogenic or likely pathogenic mutation(s) were identified in genes encoding beta-myosin heavy chain (MYH7), myosin-binding protein C (MYBPC3), cardiac troponin I and T (TNNI3 and TNNT2), tropomyosin alpha-1 chain (TPM1), myosin light chain 3 (MYL3), myosin regulatory light chain 2 (MYL2), alpha cardiac actin (ACTC1) and muscle LIM protein (CSRP3); group B – those with negative sarcomeric genetic testing, in whom pathogenic or likely pathogenic mutation(s) were identified in none of the above nine genes; and group C – those in whom genetic testing in sarcomeric genes had not been performed. Each group was then characterized according to whether exclusion of FD had been considered (and methods used in its exclusion), associated extracardiac conditions, ECG abnormalities and echocardiographic (echo) characteristics. Finally, the groups were analyzed for the presence of specific features that could potentially constitute red flags for FD. These included clinical data, specific ECG abnormalities (short or prolonged PR interval, intraventricular conduction disturbances, or the need for pacemaker [PM] implantation for treatment of bradyarrhythmias), or concentric LVH on echocardiographic imaging. Data from group B (patients with no identified sarcomeric mutations) – the particular population of interest in this study – were compared with data from groups A and C.
Statistical analysisContinuous variables are reported as means and standard deviations and comparisons were performed by ANOVA, using the F test or Welch test. The latter was used whenever the hypothesis of homogeneity of variance was rejected by Levene's test. Normality was tested by the Shapiro-Wilks test. Multiple comparisons between the two groups A and C and group B were performed using Dunnett's test or Dunnett's T3 test. Male and female ages were compared within each group using the t test. Categorical variables are expressed as absolute frequencies and percentages and differences were analyzed using Pearson's chi-square test. When the conditions for a chi-square test were not met, the Monte Carlo simulation method was used as an alternative. Male and female percentages were compared within each group by a binomial test with test percentage 50%. All p-values reported are from two-tailed tests and regarded as statistically significant at a level of 5%. Multiple comparisons were performed using Pearson's chi-square test, adjusting the significance level to 2.5% by Bonferroni's method. All data analyses were performed using IBM SPSS Statistics version 19.0.0.2.
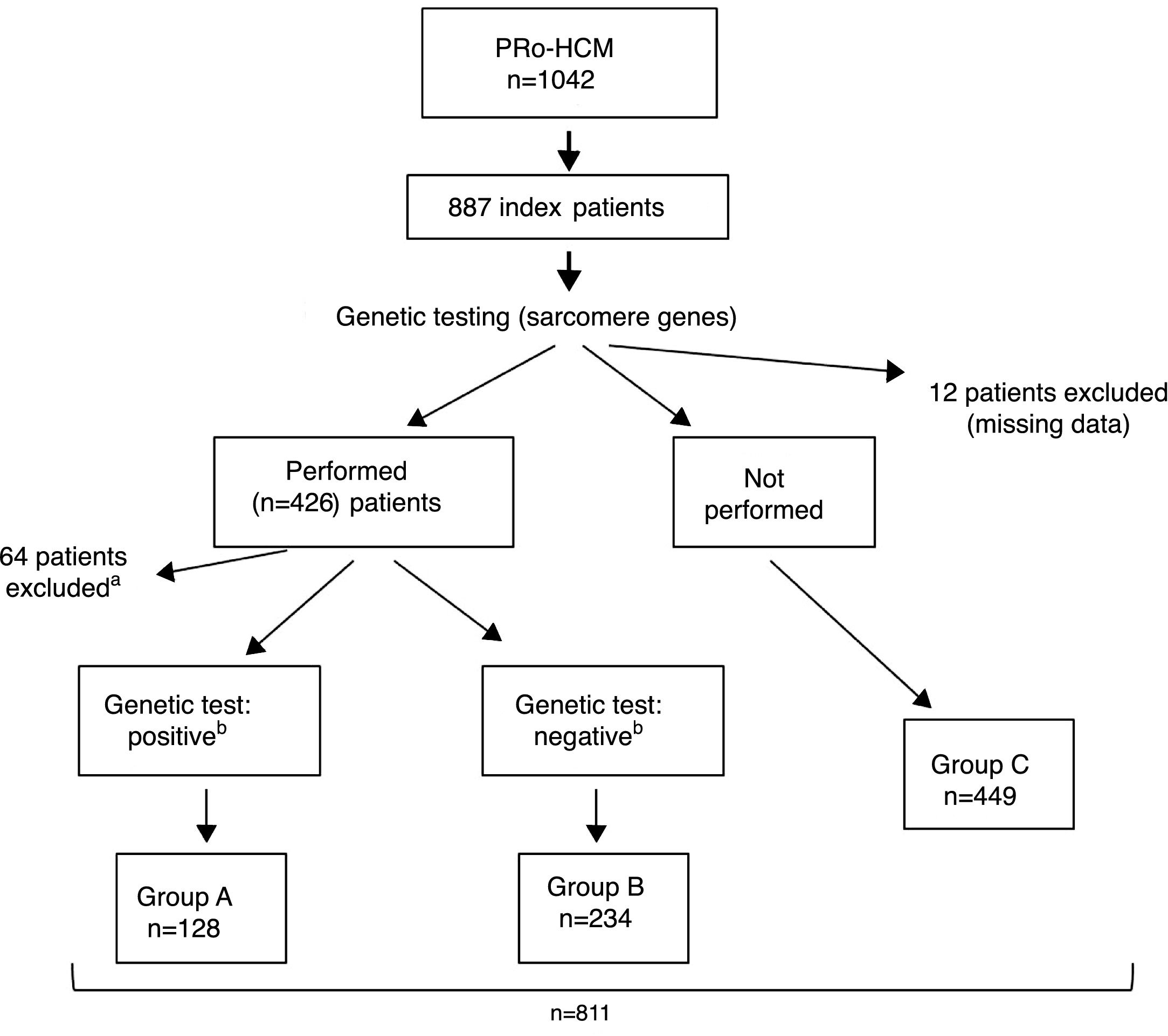
ResultsIndex patients and characteristics of study groupsThe flowchart of selection of index patients and division into groups is presented in Figure 1. A total of 1042 patients with unexplained LVH identified by imaging methods (echocardiography in 97% of cases) and a diagnosis of HCM were included in the PRo-HCM registry. Of these, 887 were index patients. Genetic testing in sarcomeric genes was performed in 48% (n = 426) of patients, and not performed in 50.6% (n = 449); 76 patients were excluded (12 due to missing data, 37 awaiting results and 27 due to unclear results). The population included in the final analysis was composed of 811 index patients.
 in sarcomeric genes encoding beta-myosin heavy chain (MYH7), myosin-binding protein C (MYBPC3), cardiac troponin I and T (TNNI3 and TNNT2), tropomyosin alpha-1 chain (TPM1), myosin light chain 3 (MYL3), myosin regulatory light chain 2 (MYL2), alpha cardiac actin (ACTC1), and muscle LIM protein (CSRP3). PRo-HCM: Portuguese Registry of Hypertrophic Cardiomyopathy.")
Flowchart of selection of index patients and division into groups for analysis and comparisons. a See text for details. b Genetic test: positive/negative: identification/non-identification of pathogenic or likely pathogenic mutation(s) in sarcomeric genes encoding beta-myosin heavy chain (MYH7), myosin-binding protein C (MYBPC3), cardiac troponin I and T (TNNI3 and TNNT2), tropomyosin alpha-1 chain (TPM1), myosin light chain 3 (MYL3), myosin regulatory light chain 2 (MYL2), alpha cardiac actin (ACTC1), and muscle LIM protein (CSRP3). PRo-HCM: Portuguese Registry of Hypertrophic Cardiomyopathy.
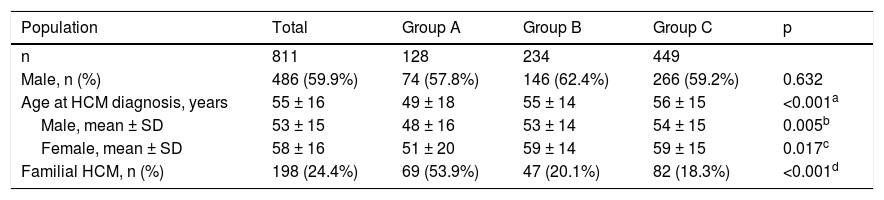
The characteristics of this population are summarized in Table 1. Males (n = 486) predominated over females (n = 325) (p<0.001); the overall median age was 55 ± 16 years at the time of diagnosis, women being older than men (p<0.001). Familial HCM was documented in 198 (24.4%) patients.
Index patients with hypertrophic cardiomyopathy included in the study.
| Population | Total | Group A | Group B | Group C | p |
|---|---|---|---|---|---|
| n | 811 | 128 | 234 | 449 | |
| Male, n (%) | 486 (59.9%) | 74 (57.8%) | 146 (62.4%) | 266 (59.2%) | 0.632 |
| Age at HCM diagnosis, years | 55 ± 16 | 49 ± 18 | 55 ± 14 | 56 ± 15 | <0.001a |
| Male, mean ± SD | 53 ± 15 | 48 ± 16 | 53 ± 14 | 54 ± 15 | 0.005b |
| Female, mean ± SD | 58 ± 16 | 51 ± 20 | 59 ± 14 | 59 ± 15 | 0.017c |
| Familial HCM, n (%) | 198 (24.4%) | 69 (53.9%) | 47 (20.1%) | 82 (18.3%) | <0.001d |
Group A: patients with identified pathogenic or likely pathogenic mutation(s) in sarcomeric genes (see text for details); Group B: patients with no identified pathogenic or likely pathogenic mutations in sarcomeric genes; Group C: patients who did not undergo genetic testing for sarcomeric genes. HCM: hypertrophic cardiomyopathy; SD: standard deviation.
Group A was composed of 128 patients (35.5% of the population in whom sarcomeric genetic testing was performed), mean age 49 ± 18 years, 74 men (48 ± 16 years) and 54 women (51 ± 20 years); group B included 234 patients, mean age 55 ± 14 years, 146 men (53 ± 14 years) and 88 women (59 ± 14 years); and group C included 449 patients, mean age 56 ± 15 years, 266 men (54 ± 15 years), and 183 women (59 ± 15 years). Although no differences were observed regarding the number of male or female patients between groups (p = NS), male gender predominated over female (p <0.001) in groups B and C, but not in group A (p = NS). Patients in group A were younger (both genders) than in the other two groups, and more frequently had familial HCM (53.9%) compared to groups B (20.1%) and C (18.3%) (p <0.001).
There was no difference between groups A and B regarding the number of sarcomeric genes tested (median 8.0, interquartile range 5-9, p = 0.051); at least five genes were sequenced in 82% of patients in group A and in 88.9% of patients in group B, and all nine genes were screened in 35.2% of patients in group A and in 34.2% of patients in group B. Screening of the MYH7, MYBPC3, MYL2, TNNI3, and CSRP3 genes was performed in the same number of patients in both groups (p = NS), but in group B (patients with no identified pathogenic or likely pathogenic mutations), four genes were screened in a larger number of patients than in group A: TNNT2 (88.9% vs. 80.5%, p = 0.028), TPM1 (70.1% vs. 58.6%, p = 0.027), MYL3 (76.9% vs. 67.2%, p = 0.045), and ACTC1 (69.2% vs. 53.1%, p = 0.002).
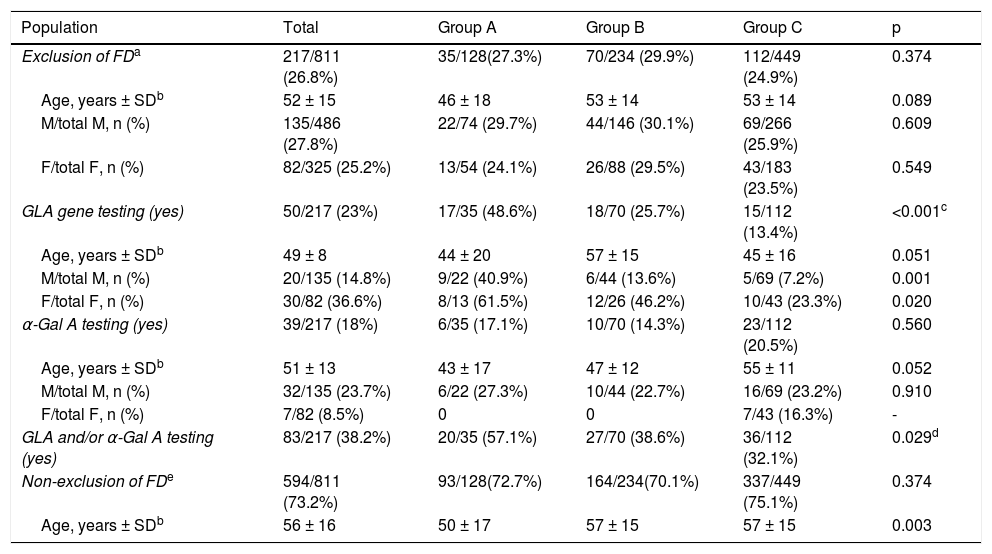
Exclusion of Fabry diseaseReported data on exclusion of FD are summarized in Table 2. Overall, FD was reported to have been excluded in 217 out of 811 patients (26.8%), in similar proportions in male (135/486; 27.8%) and in females (82/325; 25.2%), females being older than males (overall median age at diagnosis was 56 ± 15 years and 49 ± 14 years respectively; p = 0.001). There were no differences between groups regarding the number of males or females in whom exclusion of FD was reported, and the age of males was similar in all groups, as was the age of females. However, when compared within groups, females were older than males in groups B and C, but not in group A.
Exclusion of Fabry disease in index patients with hypertrophic cardiomyopathy.
| Population | Total | Group A | Group B | Group C | p |
|---|---|---|---|---|---|
| Exclusion of FDa | 217/811 (26.8%) | 35/128(27.3%) | 70/234 (29.9%) | 112/449 (24.9%) | 0.374 |
| Age, years ± SDb | 52 ± 15 | 46 ± 18 | 53 ± 14 | 53 ± 14 | 0.089 |
| M/total M, n (%) | 135/486 (27.8%) | 22/74 (29.7%) | 44/146 (30.1%) | 69/266 (25.9%) | 0.609 |
| F/total F, n (%) | 82/325 (25.2%) | 13/54 (24.1%) | 26/88 (29.5%) | 43/183 (23.5%) | 0.549 |
| GLA gene testing (yes) | 50/217 (23%) | 17/35 (48.6%) | 18/70 (25.7%) | 15/112 (13.4%) | <0.001c |
| Age, years ± SDb | 49 ± 8 | 44 ± 20 | 57 ± 15 | 45 ± 16 | 0.051 |
| M/total M, n (%) | 20/135 (14.8%) | 9/22 (40.9%) | 6/44 (13.6%) | 5/69 (7.2%) | 0.001 |
| F/total F, n (%) | 30/82 (36.6%) | 8/13 (61.5%) | 12/26 (46.2%) | 10/43 (23.3%) | 0.020 |
| α-Gal A testing (yes) | 39/217 (18%) | 6/35 (17.1%) | 10/70 (14.3%) | 23/112 (20.5%) | 0.560 |
| Age, years ± SDb | 51 ± 13 | 43 ± 17 | 47 ± 12 | 55 ± 11 | 0.052 |
| M/total M, n (%) | 32/135 (23.7%) | 6/22 (27.3%) | 10/44 (22.7%) | 16/69 (23.2%) | 0.910 |
| F/total F, n (%) | 7/82 (8.5%) | 0 | 0 | 7/43 (16.3%) | - |
| GLA and/or α-Gal A testing (yes) | 83/217 (38.2%) | 20/35 (57.1%) | 27/70 (38.6%) | 36/112 (32.1%) | 0.029d |
| Non-exclusion of FDe | 594/811 (73.2%) | 93/128(72.7%) | 164/234(70.1%) | 337/449 (75.1%) | 0.374 |
| Age, years ± SDb | 56 ± 16 | 50 ± 17 | 57 ± 15 | 57 ± 15 | 0.003 |
α-Gal A: alpha-galactosidase A activity; F: females; FD: Fabry disease; Group A: patients with identified pathogenic or likely pathogenic mutation(s) in sarcomeric genes (see text for details); Group B: patients with no identified pathogenic or likely pathogenic mutations in sarcomeric genes; Group C: patients who did not undergo genetic testing for sarcomeric genes; HCM: hypertrophic cardiomyopathy; M: males; SD: standard deviation; yes: when GLA gene testing was performed, or α-Gal A activity was assessed.
Genetic testing of the GLA gene was carried out in 50 out of 217 patients, overall more frequently in women than in men (36.6% vs. 14.8%, p < 0.001). However, GLA gene testing was performed in only 25.7% of patients in group B, with a significant difference (p = 0.019) in comparison with group A (GLA testing in 48.6% of patients), but similar to group C (13.4% of patients; p = NS); these differences between groups B and A were observed for both men (p = 0.001) and women (p = 0.020). In groups B and C, GLA gene testing was performed significantly more frequently in women than in men (respectively 46% vs. 13.6%, p = 0.003; and 23% vs. 7.2%, p = 0.016), with no age difference between genders. In group A, GLA gene testing was carried out as frequently in women as in men (61.5% vs. 40.9%, p = NS), also with no observed differences in age between genders.
In the 50 patients who underwent GLA gene testing, no pathogenic mutations were identified in 33; in 17 patients (10 in group B and 7 in group C) genetic results were missing in the registry. GLA gene screening was not performed in 74.3% of patients in group B (including in 53.8% of female patients in this group) and in 86.6% of patients in group C (including in 76.7% of female patients in this group).
Alpha-galactosidase A activityAlpha-Gal A enzyme activity was assessed in only 39/217 (18%) of patients, with no difference between groups (p = NS), and more often in men than in women (23.7% vs. 8.5%, p = 0.005). In patients with a borderline α-Gal A value, no pathogenic mutations were identified in the GLA gene. α-Gal A activity was not determined in 85.7% of patients in group B, or in 79.5% of patients in group C (≈77% of the male population in both groups).
In summary, when exclusion of FD was considered, a specific diagnostic method (GLA gene testing and/or determination of α-Gal A activity) was carried out in 38.6% of patients in group B (patients with no identified mutations in sarcomeric genes), and in 32.1% of patients in group C (patients who did not undergo sarcomeric genetic testing); in the other patients in these two groups, cardiologists relied solely on clinical and/or imaging criteria for exclusion of FD. Overall, exclusion of FD was not considered in 73.2% of the population, with no difference between groups.
Associated disorders (Supplementary Table 1)Systemic hypertension (stage 1) was common, affecting 403/811 (49.7%) of patients (mean age 62 ± 12 years at HCM diagnosis), 225/486 (46.3%) of men, and 178/325 (54.8%) of women. Hypertension was more frequent in patients in groups B (58.1%) and C (49.9%) than in group A (33.6%) (p < 0.001). Other non-cardiovascular diseases were present in 40.2% of patients overall, with no difference between groups (p = NS); the prevalence of cerebrovascular disease, renal disease, and diseases of other organs or systems was similar between groups, affecting both genders equally in all groups.
Electrocardiographic and echocardiographic dataAn abnormal ECG was present in around 95% of patients. Overall, the three groups did not differ in terms of the presence of LVH criteria (≈64%), repolarization (ST-T) abnormalities (≈82%), atrial fibrillation (≈10%), left anterior fascicular block (13%), short PR interval (3.3%), prolonged PR interval (1.4%), left bundle branch block (4.7%), right bundle branch block (10%), or need for PM implantation (6.5%) due to bradyarrhythmias or advanced atrioventricular (AV) block. Regarding gender, the groups were generally similar in the numbers of males or females with these abnormalities, although LVH criteria were more often observed in males in group B (p = 0.019 for B vs. C), and left bundle branch block was more common in females in group B (p = 0.001 for B vs. C). PM implantation was more frequently needed in older patients (mean age 63 ± 14 years, p = 0.016 between groups) (Supplementary Table 2).
A pattern of concentric LVH on echo imaging was described in 7.8% of patients, with no difference between groups. Left ventricular outflow tract or intraventricular obstruction was diagnosed in about 40% of patients, also with no difference between groups. Both of these features affected both genders equally (Supplementary Table 3).
Exclusion of Fabry disease in patients with potential red flagsA further analysis was performed comparing the three groups of patients with regard to exclusion of FD but including only those with potential red flags for the condition. These included references in the registry to prespecified clinical conditions (cerebrovascular disease, other neurological disease, or renal disease) and/or other nonspecific conditions (gastrointestinal symptoms, chronic fatigue, chronic limb pain, hearing impairment, and eye or skin problems) and/or specific ECG abnormalities (short or prolonged PR interval, intraventricular conduction abnormalities, or need for PM implantation due to bradyarrhythmias) and/or a pattern of concentric LVH on echo.
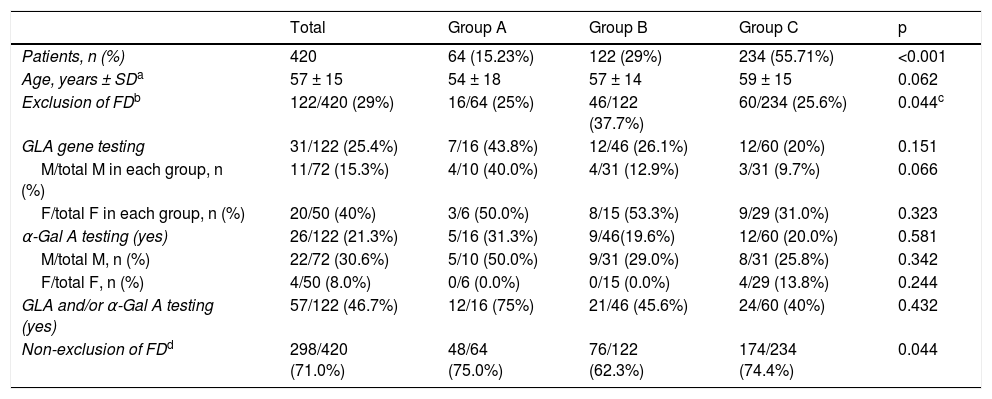
Data on exclusion of FD in patients with HCM and potential red flags are summarized in Table 3.
Exclusion of Fabry disease in patients with hypertrophic cardiomyopathy and potential red flags.
| Total | Group A | Group B | Group C | p | |
|---|---|---|---|---|---|
| Patients, n (%) | 420 | 64 (15.23%) | 122 (29%) | 234 (55.71%) | <0.001 |
| Age, years ± SDa | 57 ± 15 | 54 ± 18 | 57 ± 14 | 59 ± 15 | 0.062 |
| Exclusion of FDb | 122/420 (29%) | 16/64 (25%) | 46/122 (37.7%) | 60/234 (25.6%) | 0.044c |
| GLA gene testing | 31/122 (25.4%) | 7/16 (43.8%) | 12/46 (26.1%) | 12/60 (20%) | 0.151 |
| M/total M in each group, n (%) | 11/72 (15.3%) | 4/10 (40.0%) | 4/31 (12.9%) | 3/31 (9.7%) | 0.066 |
| F/total F in each group, n (%) | 20/50 (40%) | 3/6 (50.0%) | 8/15 (53.3%) | 9/29 (31.0%) | 0.323 |
| α-Gal A testing (yes) | 26/122 (21.3%) | 5/16 (31.3%) | 9/46(19.6%) | 12/60 (20.0%) | 0.581 |
| M/total M, n (%) | 22/72 (30.6%) | 5/10 (50.0%) | 9/31 (29.0%) | 8/31 (25.8%) | 0.342 |
| F/total F, n (%) | 4/50 (8.0%) | 0/6 (0.0%) | 0/15 (0.0%) | 4/29 (13.8%) | 0.244 |
| GLA and/or α-Gal A testing (yes) | 57/122 (46.7%) | 12/16 (75%) | 21/46 (45.6%) | 24/60 (40%) | 0.432 |
| Non-exclusion of FDd | 298/420 (71.0%) | 48/64 (75.0%) | 76/122 (62.3%) | 174/234 (74.4%) | 0.044 |
α-Gal A: alpha-galactosidase A activity; F: females; FD: Fabry disease; Group A: patients with identified pathogenic or likely pathogenic mutation(s) in sarcomeric genes (see text for details); Group B: patients with no identified pathogenic or likely pathogenic mutations in sarcomeric genes; Group C: patients who did not undergo genetic testing for sarcomeric genes; HCM: hypertrophic cardiomyopathy; M: males; SD: standard deviation; yes: when GLA gene testing was performed, or α-Gal A activity was assessed.
Four hundred and twenty index patients – 64 (15.23%) patients in group A, 122 (29.0%) in group B, and 234 (55.71%) in group C (p<0.001 for comparison between groups) – had at least one of the above features. In this sub-analysis, exclusion of FD was reported in 122 (29%) patients, more frequently in group B patients with these features compared to group C (37.7% vs. 25.6%, p = 0.018), but with no difference compared to group A (25%, p = NS). However, exclusion of FD relied on GLA gene testing in only 25.4% of patients, with no difference between groups (p = NS), more commonly in females than males in groups B and C; and α-Gal A activity was assessed in 21.3% of patients, also with no differences between groups (p = NS), and mostly in men.
Therefore, in patients with red flags for FD and in whom the disease was reported as having been excluded, a specific test was performed in 46.7% of patients (including 45.6% of those in group B and 40% of those in Group C); in the other patients exclusion of FD relied solely on clinical and/or imaging criteria.
Overall, exclusion of FD was not considered in 298 (71%) patients with red flags for FD, with no difference between groups (p = NS).
DiscussionIn this descriptive, observational analysis of a subpopulation of 811 index patients included in the PRo-HCM registry with a diagnosis of HCM on the basis of classic criteria,37,40,41 genetic testing in sarcomeric genes was performed in ≈45% of patients, and a pathogenic or likely pathogenic mutation associated with HCM was identified in 35.5%.
A definitive diagnosis of sarcomeric HCM can only be made when a pathogenic or likely pathogenic mutation associated with the disease is identified. However, when genetic testing targeting the most common mutated genes is performed, findings are negative in up to 50% of cases.1 It may be hypothesized that older patients with a late HCM phenotype may have mutations in other (uncommon) sarcomeric genes that are not usually screened, but it is also possible that FD is the cause of that phenotype.
According to data from the registry, exclusion of FD was infrequent and relied on specific diagnostic tests in less than 40% of patients. When sarcomeric genetic testing was negative or was not performed, consideration of FD predominantly involved older women. Clinical presentation of FD may occur later and be more variable in women than in men, and women are more likely to develop a variant characterized by isolated cardiac involvement only.6–8,10 Interestingly, the GLA gene was most often tested in patients with identified sarcomeric mutations, probably because a large panel of genes was used; but it was the only gene tested in 13% of patients in group C, possibly due to economic restraints.
However, genetic testing for FD was not performed in more than 50% of female patients in group B, or in more than 70% of females in group C. Furthermore, α-Gal A activity was only assessed in a minority of patients, although, appropriately, mainly in males; but it was not assessed in the majority of men in groups B and C. Overall, specific tests for FD exclusion were performed in less than 50% of patients in whom the disease was reported as having been excluded. GLA testing results, when available, were negative.
These data support the conclusion that many cardiologists rely only on clinical or imaging data for exclusion of FD, and consider that in most patients with an HCM phenotype specific tests are not indicated or necessary. However, this may be a misconception. In patients with no male-to-male transmission of unexplained LVH, FD should be considered as a potential diagnosis and specific tests need to be performed, a decision supported by any other additional personal and/or family feature suggesting the diagnosis.35 Needless to say, in the absence of a family history of FD, such features in family members (kidney disease, acroparesthesias, cornea verticillata, etc.) may be absent, difficult to ascertain, or simply overlooked in patients with a predominantly or exclusively cardiac phenotype.
The ECG abnormalities observed in this analysis are commonly described in patients with LVH, whatever the cause, patients with sarcomeric HCM or FD included. A short PR interval is classically described in 21%-40% of patients with FD,42 particularly in its early stages but, although less common, it is also described in HCM (3.3% in the study population). Likewise, a tendency for PR prolongation and AV block is described in the later stages of FD,43 and these abnormalities may also occur during the natural history of sarcomeric HCM. They were found with similar prevalence in all groups of the study population. Conduction abnormalities may always be considered an additional warning for the cardiologist when excluding FD, but they are in no way a decisive diagnostic marker of FD.
All echo patterns of LVH can be observed in sarcomeric HCM.44–46 Concentric LVH (the most common form in FD) was described in less than 10% of patients in the study population. Conversely, obstruction (infrequent in FD) was diagnosed in about 40% of patients. Both echo features were observed similarly, whether or not sarcomeric genetic testing had been carried out, and regardless of its result, but none of them is key to the diagnosis.
Finally, in this population there was no clear evidence of non-cardiac features suggesting FD, but symptoms or signs may be subtle and non-specific in non-classical forms. The subpopulations studied were relatively elderly for sarcomeric HCM and potentially prone to what is termed Fabry cardiomyopathy – despite the rarity of this disease – particularly considering the female gender and the forms that can be expressed with a late and predominantly cardiac phenotype.13,20
The findings herein highlight the need to be alert for both conditions: sarcomeric HCM (the purpose of the PRo-HCM registry), which is often diagnosed late as it frequently evolves without symptoms; and FD, which needs to be systematically considered in the differential diagnosis of most cases of LVH, especially when the proband is negative for sarcomeric mutations.
In the genetic era, it seems reasonable to include GLA in the panel of genes to be tested when HCM is investigated. When genetic testing is not possible, α-Gal A activity should be assessed, because it may be diagnostic in male patients, with GLA testing being reserved as a second step if appropriate. In females, particularly if middle-aged or older and bearing in mind the possibility of a predominantly cardiac phenotype, GLA gene testing is advisable even if it is the only gene that can be tested due to economic constraints or other reasons.
LVH occurs late in FD, but enzyme replacement treatment can favorably modify the long-term prognosis of the disease, reducing the incidence of and/or progression to serious clinical events,3,4,31,32 and when a diagnosis is made in the index patient, early diagnosis in affected relatives is facilitated47 and may be associated with better outcomes.
LimitationsThe PRo-HCM registry was designed for the study of patients with a previous diagnosis of sarcomeric HCM on the basis of classical criteria, and not according to a genetic diagnosis. It cannot be excluded that patients with no identified pathogenic mutations in common sarcomeric genes have HCM associated with mutations in other rarer genes that are not usually screened. But it also cannot be ruled out that in fact some of the cases of HCM included in the registry are atypical FD with a late expression and a predominantly or exclusively cardiac phenotype.
We considered as potential red flags for FD clinical features that were not fully characterized in PRo-HCM, because the registry was not designed for investigation of unexplained LVH. However, considering the similarities in clinical, ECG and echocardiographic profile observed in the three patient groups, rare genetic diseases such as FD seem unlikely as a cause of HCM in this population. Also, systemic hypertension was a common finding, as expected considering its prevalence worldwide and the age of the population,48,49 although it is improbable that stage 1 hypertension could explain the observed ≥15 mm LVH on echo imaging.
However, the aim of this descriptive analysis, which we consider original in design and objectives, was neither to assess the yield of genetic testing in sarcomeric HCM nor to determine the prevalence of FD in patients with unexplained LVH detected by imaging techniques, but only to assess the awareness of cardiologists of a potential diagnosis of FD, and in clinical practice this awareness seems to be lower than empirically suspected.
ConclusionsAnalysis of data from subpopulations of patients included in the PRo-HCM registry revealed a low awareness among cardiologists of the need to exclude FD in patients without a genetic diagnosis of sarcomeric HCM. When exclusion of FD was undertaken, it relied mainly on clinical or imaging features, with specific diagnostic tests performed in fewer than 50% of cases in which they could hypothetically be justified. These data, in a cardiology setting, should be taken as an indication for a broader approach to patients with unexplained HCM, aimed at the exclusion of a disease that, although rare, may benefit from specific therapy that has a favorable impact on prognosis.
FundingThe Portuguese Registry of Hypertrophic Cardiomyopathy was supported by the following companies (in alphabetical order): Jaba Recordati, Medinfar, Merck Serono, Sanofi Genzyme, Servier, Shire Human Genetic Therapies.
This article was sponsored by a grant from Sanofi Genzyme.
Authors’ contributionsDB, NC, and LR-L conceived and designed the research, and drafted the manuscript; AB performed the statistical analysis; JM, LG, and HM performed critical revisions of the manuscript for drafting and key intellectual content. All authors contribute to revising the paper and gave final approval of the version to be published.
Conflicts of interestDB reports consultancy and lecture fees from Sanofi Genzyme, and attended meetings sponsored by Shire Human Genetic Therapies; LRL attended meetings sponsored by Shire Human Genetic Therapies and Sanofi Genzyme; the other authors have no conflicts of interest to declare.
We thank the Portuguese Society of Cardiology's National Center for Data Collection in Cardiology (CNCDC) team: Sandra Corker, Adriana Belo, Lino Gonçalves (Head of CNCDC) and Jorge Mimoso, and investigators from the participating centers (see Appendix A).
Centro Hospitalar de Leiria - Joana Correia (PI), Catarina Ruivo, Fernando Sá; Centro Hospitalar de Lisboa Norte, Hospital de Santa Maria, Lisbon - Dulce Brito (PI), Ana Rita Francisco, Tatiana Guimarães, Miguel Nobre Menezes, Oana Moldovan, Mónica Mendes Pedro, Gustavo Lima da Silva; Centro Hospitalar de Lisboa Ocidental, Serviço de Cardiologia - João Abecasis (PI), Francisco Moscoso Costa, Helder Dores; Centro Hospitalar de Lisboa Ocidental, Hospital São Francisco Xavier, Serviço de Medicina III: Cândida Fonseca (PI), Inês Araújo, Filipa Marques; Centro Hospitalar de Trás os Montes e Alto Douro, Hospital de São Pedro, Vila Real - Carla Alexandra R. Araújo (PI), Ana Isabel Baptista, Sofia Silva Carvalho, Filipa Cordeiro, Catarina Ferreira, Sílvia Leão, Pedro Magalhães, Renato Margato; Centro Hospitalar de Vila Nova de Gaia/Espinho - Conceição Fonseca (PI), João Tiago Almeida, Ricardo Fontes-Carvalho, Rita Faria, Paulo Fonseca, Olga Sousa; Centro Hospitalar do Algarve, Hospital de Faro - Nuno Marques (PI), José Amado, Joana Chin, Walter Santos; Centro Hospitalar do Alto Ave, Hospital da Senhora da Oliveira, Guimarâes - Olga Azevedo (PI), Margarida Oliveira, Lucy Calvo, João Português; Centro Hospitalar do Baixo Vouga, Hospital Infante D. Pedro, Aveiro - José António Nobre dos Santos (PI), Tiago Cardoso, Ana Raquel Ferreira, José Luis Martins; Centro Hospitalar do Oeste Norte, Centro Hospitalar das Caldas da Rainha - Ana Filipa Pereira Rodrigues (PI); Centro Hospitalar do Porto, Hospital de Santo António - Patrícia Fernandes Rodrigues (PI), Paulo Palma, Mário Santos, Maria João Monteiro e Sousa; Centro Hospitalar do Tâmega e Sousa, Unidade Padre Américo, Penafiel - Maria Conceição Queirós (PI), Alexandra Castro, Adriana Pereira; Centro Hospitalar e Universitário de Coimbra, Cardiologia B, Hospital Geral - Joana Delgado Silva (PI), Ana Botelho, Liliana Reis Teles; Centro Hospitalar Tondela Viseu, Hospital de São Teotónio - Carlos Emanuel Correia (PI), Luis Abreu, Davide Moreira; CUF Infante Santo Hospital, Lisbon - Pedro Matos (PI); Hospital Beatriz Ângelo, Loures - Luis Sargento (PI), Miguel Almeida Ribeiro; Hospital da Luz, Lisbon - Nuno Cardim (PI); Hospital das Forças Armadas, Lisbon - Sara Ferreira (PI); Hospital de Braga: Nuno Salomé (PI), Carina Arantes, Carlos Braga, António Costeira, Catarina Vieira; Hospital de Santa Maria Maior de Barcelos, Serviço Cardiologia - Alexandra Sousa (PI), Mariana Paiva; Hospital de Santo Espírito de Angra do Heroísmo, Açores - Rute Couto (PI); Hospital de São João, Porto - Elisabete Martins (PI), Ana Lebreiro, Sérgio Leite, Carla Sousa; Hospital do Espírito Santo, Évora - Agostinho Caeiro (PI), João Filipe Carvalho, Bruno Piçarra; Hospital Garcia de Orta, Almada - Luis Rocha Lopes (PI), Ana Rita Almeida, Inês Cruz; Hospital Prof. Doutor Fernando Fonseca, Amadora - Francisco Madeira (PI), Mariana Faustino; Hospital SAMS, Lisbon - Berta Carôla (PI), Rui Conduto, Paulo Pedro; HPP Hospital de Cascais, Hospital Dr. José de Almeida - Gonçalo Proença (PI), Élia Batista, Sara Eira, Carla Matias; Unidade Local de Saúde da Guarda, Hospital Sousa Martins - Maria Cristina Gamboa (PI), Maria Isabel Santos.
Centers are in alphabetical order. PI: Principal Investigator.
The following are the supplementary material to this article:
