
A 67-year-old man was admitted to our hospital after episodes of syncope preceded by malaise and diffuse neck and chest discomfort. No family history of cardiac disease was reported. Laboratory workup was within normal limits, including D-dimers, serum troponin I and arterial blood gases. The electrocardiogram showed sinus rhythm with T-wave inversion in leads V1 to V3. Computed tomography angiography to investigate pulmonary embolism showed no abnormal findings. Transthoracic echocardiography (TTE) displayed massive enlargement of the right ventricle with intact interatrial septum and no pulmonary hypertension. Cardiac magnetic resonance imaging (MRI) confirmed right ventricular (RV) dilatation and revealed marked hypokinesia/akinesia of the lateral wall. Exercise stress testing was negative for ischemia.
According to the 2010 Task Force criteria for arrhythmogenic right ventricular dysplasia (ARVD), this patient presented two major criteria (global or regional dysfunction and structural alterations: by MRI, regional RV akinesia or dyskinesia or dyssynchronous RV contraction and RV ejection fraction ≤40%, and repolarization abnormalities: inverted T waves in right precordial leads [V1, V2, and V3]); and one minor criterion (>500 ventricular extrasystoles per 24 hours by Holter), and so a diagnosis of ARVD was made.
After electrophysiologic study (EPS) the patient received an implantable cardioverter-defibrillator (ICD).
This late clinical presentation of ARVD highlights the importance of TTE screening, possibly complemented by MRI. The associated risk of sudden death was assessed by EPS leading to the implantation of an ICD. Genetic association studies should be offered to the offspring of all ARVD patients.
Um homem de 67 anos deu entrada no hospital após episódios de síncope precedidos por mal-estar geral e desconforto torácico difuso. Sem história familiar de doença cardíaca. O estudo analítico, incluindo d-dímeros e troponina I, bem como a gasimetria arterial, não mostrou alterações. O eletrocardiograma exibiu um ritmo sinusal com inversão da onda T de V1-V3. A angioTC foi negativa para tromboembolismo pulmonar.
O ecocardiograma transtorácico (ETT) mostrou uma dilatação marcada do ventrículo direito com septo interauricular intacto, sem hipertensão arterial pulmonar.
A ressonância magnética cardíaca (RMC) confirmou a dilatação ventricular direita e revelou hipocinesia marcada/acinesia da sua parede lateral. A prova de esforço foi negativa para isquemia.
À luz dos critérios de diagnóstico de displasia arritmogénica do ventrículo direito (DAVD), preconizados pela Task Force de 2010, este doente apresentava dois critérios major (na RMC: acinesia ou discinesia regional do VD e fração de ejeção≤40%; no eletrocardiograma: ondas T invertidas nas derivações pré-cordiais direitas [V1-3]) e um minor (no ECGD-Holter de 24 horas>500 extrassístoles ventriculares), pelo que o diagnóstico definitivo foi assumido.
Após o estudo eletrofisiológico, o doente foi submetido a implantação de cardioversor-desfibrilhador (CDI) monocâmara, encontrando-se assintomático desde então.
Esta apresentação tardia de DAVD evidencia a importância do rastreio por ETT, complementado por RMC. O risco de morte súbita cardíaca foi aferido pelo estudo eletrofisiológico, conduzindo à implantação do CDI. Estudos de associação genética devem ser oferecidos aos descendentes do paciente.
A 67-year-old man, independent in daily activities, a former pilot, with a history of essential hypertension and dyslipidemia (both under therapy), a current smoker (75 pack/years), and with no relevant family history, went to an emergency department due to an episode of general malaise accompanied by a sensation of constriction at the front of the neck and slight chest discomfort, followed by sudden loss of consciousness with complete and immediate spontaneous recovery. He reported no typical constricting chest pain, dyspnea, sweating, nausea or vomiting, asthenia, lower limb edema, focal neurologic signs, seizures or sphincter incontinence. There had been no further symptoms since that episode other than a feeling of general malaise.
Laboratory workup revealed negative troponin I and D-dimers. Arterial blood gases, posteroanterior chest X-ray, electrocardiogram (ECG), computed tomography (CT) angiography and brain CT showed no pathological alterations.
According to the patient, a provisional diagnosis of acute coronary syndrome was initially made and he was kept under observation for 24 hours. He was discharged the following morning without a definitive diagnosis.
The patient came to our emergency department 24 hours later with the same clinical setting except without loss of consciousness. He also reported a few seconds of palpitations. He denied other symptoms, and mentioned similar episodes a few weeks previously.
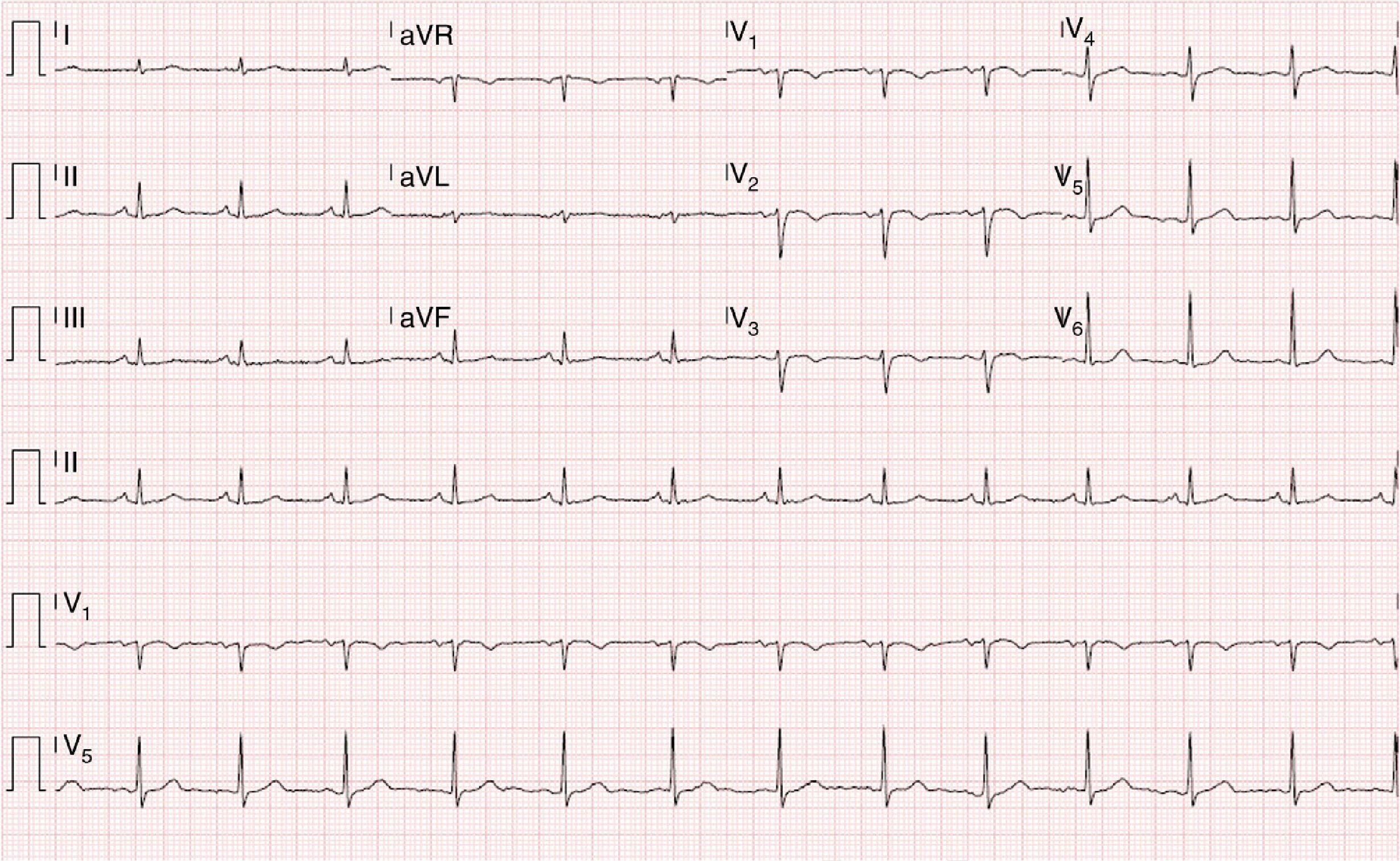
Physical examination was normal. The ECG showed sinus rhythm with no signs of acute ischemia, T-wave inversion in V1-V3 and absence of epsilon waves (Figure 1).

The posteroanterior chest X-ray showed no relevant alterations.
Blood tests revealed no abnormalities with no elevation of acute phase parameters, and renal function, electrolytes, coagulation tests, troponin I and CK-MB were within normal limits.
Transthoracic echocardiography (TTE) showed an undilated left ventricle with good global systolic function, no wall motion abnormalities and ejection fraction (EF) of 64%; massive right ventricular (RV) dilatation, with tricuspid annular plane systolic excursion (TAPSE) of 19 mm, dilated right atrium, <50% inspiratory collapse of the inferior vena cava and pulmonary artery systolic pressure of 31.60 mmHg; and no other relevant findings.
During hospital stay the patient's symptoms disappeared. Laboratory tests showed NT-proBNP of 705 pg/ml but no other abnormalities.
Given the unexplained loss of consciousness together with the presence of RV dilatation with no apparent alterations in wall motion or ventricular function, 24-hour Holter ECG monitoring, exercise testing and cardiac magnetic resonance imaging (MRI) were requested.
Holter monitoring revealed a mean of 22 isolated bimorphic ventricular extrasystoles per hour (528 in 24 hours), and six pairs. Exercise testing using the Bruce protocol had a duration of 7 min 30 s and was terminated by patient fatigue, at which point he had reached 90% of age-predicted maximum heart rate. During the test the patient reported no angina and no pathologic hemodynamic or electrocardiographic alterations were observed.
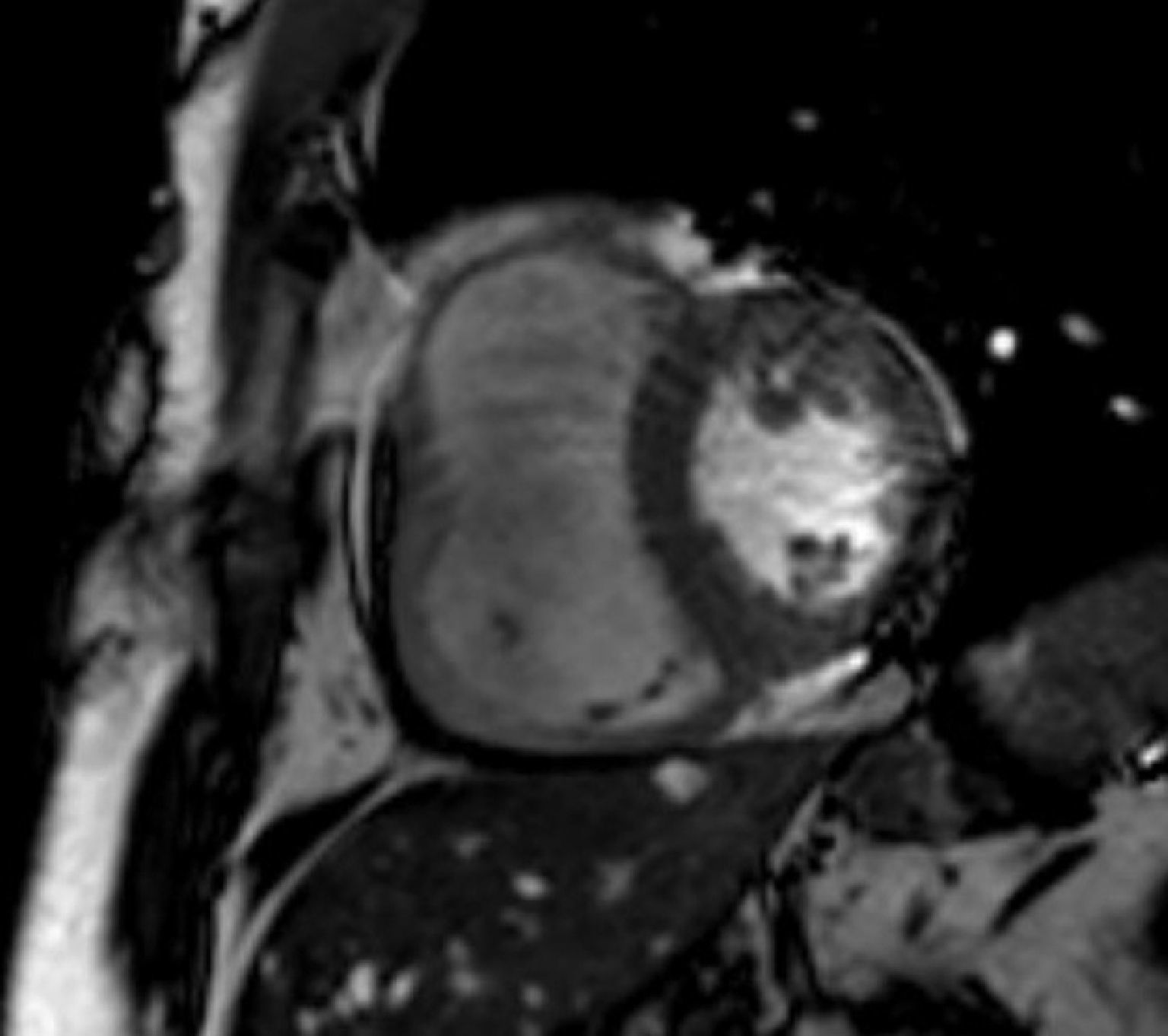
MRI revealed a dilated right ventricle with marked hypokinesia/akinesia of the free wall and EF of 21%. No late enhancement was seen in the myocardium after injection of paramagnetic contrast (Figure 2).

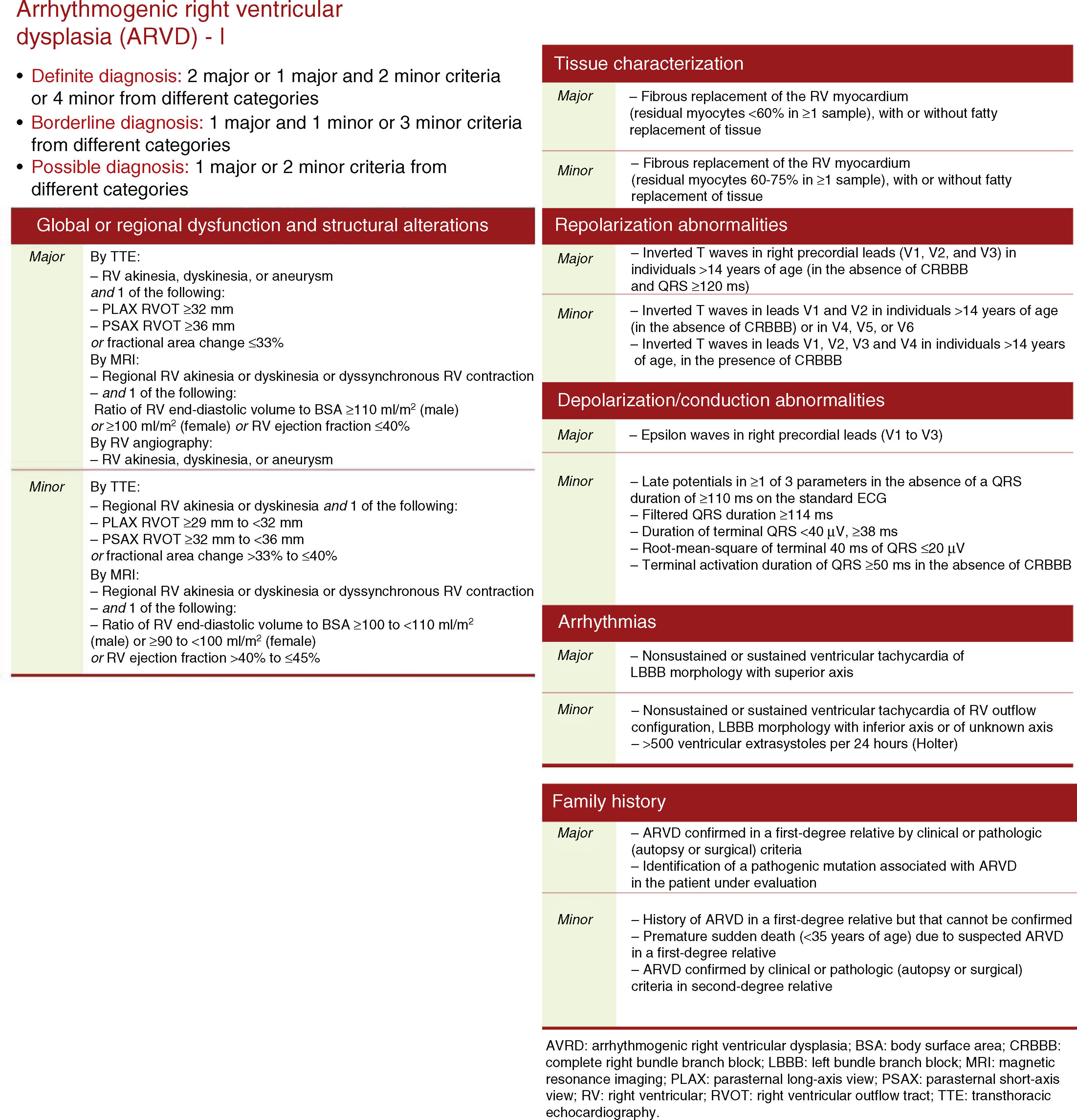
On the basis of these findings, the patient was judged to meet two major and one minor criteria for a definitive diagnosis of arrhythmogenic right ventricular dysplasia (ARVD) according to the 2010 Task Force1 (by MRI: regional RV akinesia or dyskinesia or dyssynchronous RV contraction and RV ejection fraction ≤40%; on ECG: inverted T waves in right precordial leads [V1-V3]; and >500 ventricular extrasystoles per 24 hours by Holter) (Figure 3).

Diagnostic criteria for arrhythmogenic right ventricular dysplasia. • Definite diagnosis: 2 major or 1 major and 2 minor criteria or 4 minor from different categories. • Borderline diagnosis: 1 major and 1 minor or 3 minor criteria from different categories. • Possible diagnosis: 1 major or 2 minor criteria from different categories.
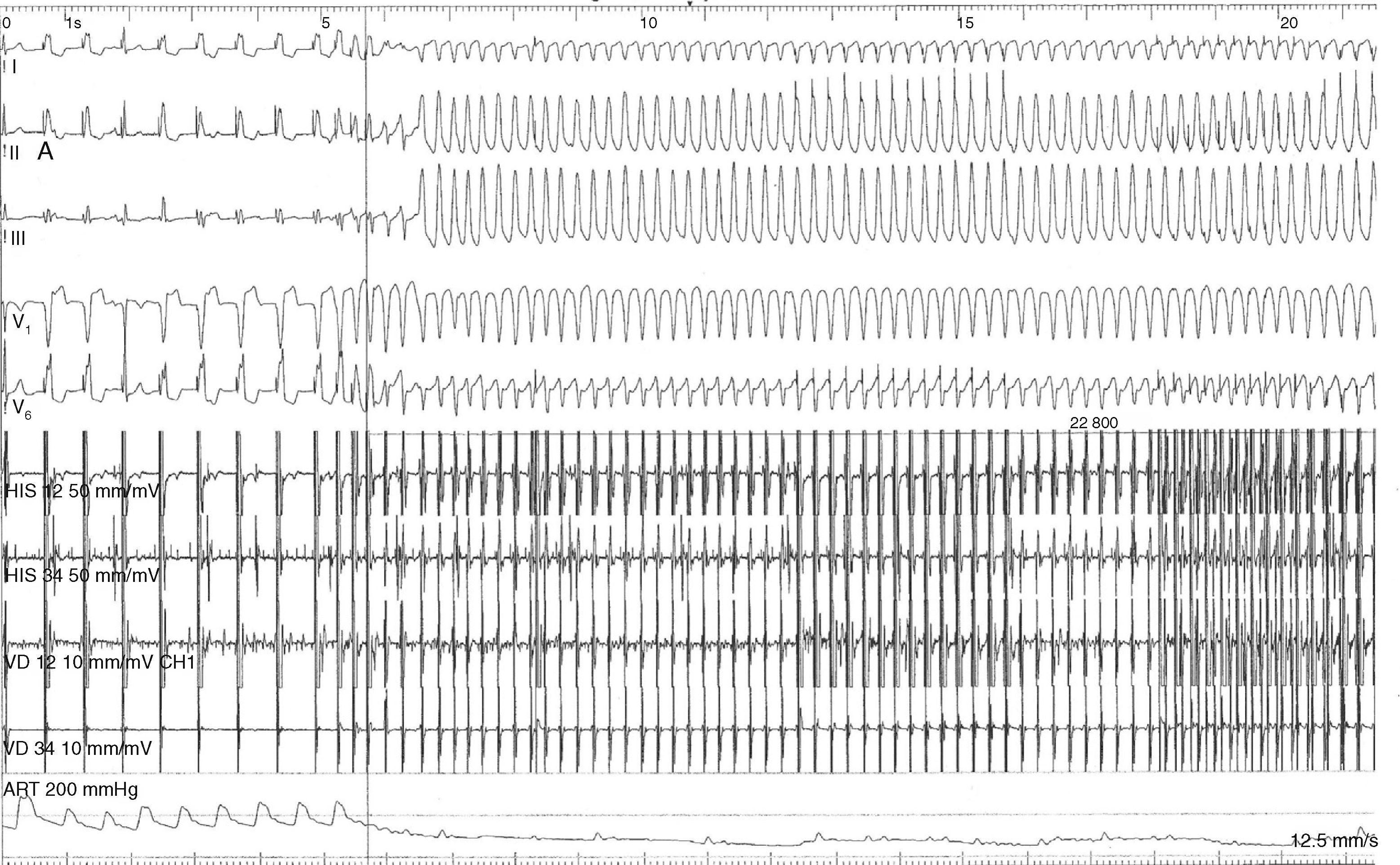
To assess the risk of sudden death, electrophysiologic study (EPS) was performed, which documented inducible monomorphic ventricular tachycardia (VT) with complete left bundle branch block morphology and inferior axis, resulting in hemodynamic collapse on two occasions, one requiring electrical cardioversion to a polymorphic form and the other terminated by antitachycardia pacing (Figure 4).

The patient received a single-chamber implantable cardioverter-defibrillator (ICD), which was implanted without complications.
The patient was advised to limit physical exertion and was prescribed beta-blockers (bisoprolol 2.5 mg daily) in addition to his usual medication.
He remained asymptomatic and tolerated the ICD well. Genetic study and electrocardiographic and echocardiographic monitoring have been recommended for his offspring.1,2
DiscussionEpidemiologyThere are limited epidemiologic data on ARVD. Its prevalence in the general population is between 1/2000 and 1/5000, with males being affected more often than females (3:1). Its incidence ranges between 1/1000 and 1/50 000, with considerable geographic variability.3–5
In most cases (80%) ARVD is diagnosed before the age of 40. Worldwide, it is identified as the cause of sudden cardiac death in young adults in 5-11% of cases. In a study in northern Italy it was the leading cause (22.4%) of sudden death in young athletes.6
ARVD should therefore be considered if previously healthy young individuals present with arrhythmia, syncope or cardiac arrest. An initial episode at older ages is unusual, and diagnosis of late-onset ARVD is made more difficult by possible confounding factors such as concomitant coronary artery disease.7
BiopathologyElectron microscope studies8 reveal alterations in desmosomal proteins to be the main ultrastructural factor that triggers failure of myocytes to adhere, resulting in cell death and progressive replacement by fibrofatty tissue, which is the typical pathologic substrate of ARVD. These structural changes increase mechanical stress on the ventricular myocardium with focal dilatation of the chamber, initially in the thinner parts of the right ventricle (apex, inflow tract and outflow tract, known as the triangle of dysplasia), and subsequently affecting the entire ventricle. This may explain why the right ventricle, which is more distensible than the left ventricle because of its thinner wall and asymmetric shape, is more often involved in ARVD, especially in the earlier stages.9
However, LV involvement is common (>50%) and increasingly prevalent at older ages as the disease advances. In some forms of presentation LV involvement predominates, and the final expression of the disease can resemble dilated cardiomyopathy.10–12
In the initial stages, structural changes are confined to the ‘triangle of dysplasia’ and the patient will be asymptomatic, but even so may be at risk for sudden death, particularly during physical exertion. Later, the myocardial fibers in areas undergoing fibrofatty transformation are the substrate for the development of symptomatic ventricular arrhythmias, and changes in RV function become evident. The disease progresses from epicardium to endocardium and eventually spreads to the entire right ventricle, leading to marked dilatation and dysfunction.10
In the case presented, the initial episode of syncope in a previously asymptomatic 67-year-old with marked structural and functional abnormalities (severe RV dilatation with regional hypokinesia/akinesia and EF of 21%), is a good example of how ARVD can be silent even in its advanced stages.
GeneticsEstimates of the proportion of hereditary cases of ARVD vary widely, from 30% to over 50%.3,13
The main desmosomal proteins altered in ARVD are plakoglobin, desmoplakin, plakophilin-2, desmoglein-2 and desmocollin-2. Mutations in other genes have been identified, such as TMEM43 (which is associated with a high-risk form of ARVD and appears to be related to a PPAR-γ-regulated lipogenesis pathway), TGF-β3 and RYR2, the gene coding for the cardiac ryanodine receptor. The pathogenic mechanisms leading to ARVD associated with these genes are still under investigation, as are other mutations that may also be involved.1,14,15
Two forms of transmission have so far been identified, autosomal dominant (more common and with incomplete penetrance) and autosomal recessive, in which ARVD is part of a syndrome that includes palmoplantar keratoderma and woolly hair (Naxos and Carvajal syndromes, associated with mutations in the genes for plakoglobin and desmoplakin, respectively).16,17
The pattern of incomplete penetrance means that the age at which symptoms and clinical manifestations first appear varies between individuals with the same mutation and that asymptomatic carriers are at risk of developing the disease at any time.9,18
Natural history and clinical manifestationsThe natural history of ARVD depends on the rate of progression of RV dysfunction and development of symptoms.
The form of initial presentation varies widely, from palpitations, dizziness, general malaise, chest pain and dyspnea to syncope and occasionally sudden death. Symptoms usually appear between the first and fifth decades of life; mean age at diagnosis is 30 years, rarely before the age of 12 or after the age of 60.
Overall mortality is 4-20% in both sexes, peaking in the fourth decade of life, while the annual rate of sudden death is 1%. The high mortality from ARVD described in some studies, together with its progressive nature, mean that it is extremely important to identify the condition and to begin treatment promptly.3,9,15,19
A study in which hearts were examined macroscopically and microscopically after autopsy revealed that out of 1930 cases of unexpected sudden cardiac death, around 10% showed evidence of ARVD; most of these 200 deaths occurred at home, at work or in the street and only 3.5% during physical exercise.20 However, it has been demonstrated that in certain populations, exercise is an important cause of sudden death in athletes.
As stated above, even though in many patients AVRD can remain clinically silent for decades, the risk of sudden cardiac death, although relatively low, must be borne in mind, and so the diagnostic process must be undertaken without delay. However, this is not always the case, especially in sporadic cases with no known family history.21
Diagnostic approachNo single diagnostic method is conclusive, particularly in the early stages of the disease. When ARVD is suspected, the essential diagnostic exams are the ECG and TTE.22
ElectrocardiogramWhen ARVD is suspected, the ECG should always be part of the initial diagnostic approach, since abnormalities are observed in 90% of those affected.4
Although the sensitivity of the initial ECG is low (40-50% for the first episode), the sensitivity and specificity of long-term monitoring with serial ECGs are high. The most important ECG findings in patients with ARVD are repolarization abnormalities, of which T-wave inversion in leads V1-V3 is the most common (54-100% of patients). Although this alteration is one of the major criteria of the 2010 Task Force for a diagnosis of AVRD, it is not specific to this condition and can be seen in healthy individuals. Epsilon waves, found in 30% of cases and caused by delayed electrical activation of the right ventricle, are the most specific feature of AVRD. Other findings include complete or incomplete right bundle branch block and terminal QRS prolongation.23
EchocardiographyTTE is the most common method for detecting functional and structural abnormalities in the heart. It is non-invasive and readily available in most hospitals.3,4,18 However, the retrosternal position and complex geometry of the right ventricle mean it cannot be used as the only imaging method in AVRD.24
The alterations most often identified are RV dilatation, particularly of the outflow tract (there may also be regional aneurysms and atrial enlargement), morphological irregularity (found in up to 62% of cases and including trabecular derangement and hyper-reflective moderator band), and reduced right ventricular fractional area change (RVFAC), which correlates with EF. Regional wall motion abnormalities are observed in up to 80% of cases.25
The echocardiographic measures in the 2010 Task Force criteria (Figure 3) are less sensitive and hence less reliable than those of MRI, mainly due to difficulty in assessing regional ventricular wall motion abnormalities. New three-dimensional techniques look set to overcome these limitations. Studies have shown that besides its diagnostic capacity, echocardiography also has prognostic value in ARVD, demonstrating that reduced TAPSE and RVFAC are associated with major cardiac events. However, as pointed out above, major arrhythmic events can occur before the development of systolic dysfunction, and so risk stratification is hampered by the considerable variability in phenotypic expression of the disease.24
Cardiac magnetic resonance imagingAs stated above, echocardiography is not always sufficient for a diagnosis of ARVD, and MRI overcomes some of its limitations. It is a non-invasive method of detecting fibrofatty replacement of myocardium, wall thinning and regional wall motion abnormalities. The most important MRI findings for a diagnosis of ARVD are high signal intensity in the ventricular wall, which indicates fatty deposits, dilatation of the RV outflow tract, and hypokinesia, akinesia or dyskinesia and dilatation of the right ventricle and atrium.26
One MRI technique that correlates well with histopathologic findings, degree of RV dysfunction and inducibility of VT on EPS is late enhancement following injection of paramagnetic contrast to detect fibrous tissue, which was positive in 67% of patients with ARVD.27
Some studies show that MRI has greater diagnostic value than conventional echocardiography in ARVD.22,28 However, it also produces false positives, particularly if the criteria used are based solely on fibrofatty alterations and ventricular wall thickness.18,24,29
In addition to false positives, other limitations of MRI are interobserver variability in interpretation of its findings, artefacts caused by arrhythmias, restrictions to its use in the presence of intracardiac devices, cost, and lack of availability in less specialized centers.30
Holter electrocardiographic monitoringSeveral studies have demonstrated the value of Holter ECG monitoring in AVRD, to detect both ventricular extrasystoles and potentially life-threatening ventricular arrhythmias, since a correlation has been shown between the two.31–33
Electrophysiologic studyThere are conflicting data concerning programmed ventricular stimulation and VT induction for risk stratification in ARVD. Nevertheless, although induction of VT does not predict future arrhythmic events, it does identify patients at greater risk for disease progression and sudden death.34
In a study of 62 patients diagnosed with ARVD according to the 2010 Task Force criteria, 55% presented inducible monomorphic VT, and after 10 years of follow-up cardiac death, heart transplantation, and VT with hemodynamic compromise were more frequent in these patients.18,35
The 2015 update of the Task Force consensus document recommends that EPS should be considered in the diagnosis and/or evaluation of patients with suspected ARVD.36
Genetic testingGenetic testing is not recommended for all patients with suspected AVRD. In particular, while it can be useful for patients satisfying Task Force diagnostic criteria for AVRD if clinical, ECG and imaging findings are doubtful but the diagnosis is possible, it is not considered necessary (class IIb recommendation).37
Discovery of the role of TMEM43, mentioned above, has opened up new avenues for the use of genetic testing in risk stratification, since mutations in this gene have been reported to be associated with 50% mortality in males by the age of 39.38
Diagnosis and risk stratificationThe main causes of failure to diagnose ARVD are misinterpretation of MRI findings and unawareness of the 2010 Task Force criteria. In the case presented, diagnosis was based on these criteria, which update those of the 1994 document and include alterations in RV structure and function, histopathology, repolarization and depolarization abnormalities on the ECG, arrhythmias, and family history.1 The case under discussion presented two major criteria (by MRI, RV akinesia and RV ejection fraction ≤40%; and by ECG, inverted T waves in right precordial leads [V1-V3]) and one minor criterion (>500 ventricular extrasystoles per 24 hours by Holter ECG), leading to a definitive diagnosis.
A study of the diagnostic performance of imaging methods found that only 50% of patients diagnosed with ARVD by MRI fulfilled the echocardiographic criteria of the 2010 Task Force, highlighting the importance of MRI in the diagnosis of this disease.39
Of the indicators currently used for risk stratification, left and right ventricular dysfunction, mutations in TMEM43, T-wave inversion in leads V1-V3 and documented VT are the most convincing. Given the progressive nature of the disease, risk stratification should be considered a dynamic process that is subject to continual reassessment.34
Therapeutic approachTreatment should be directed primarily toward prevention of sudden cardiac death, but the best way to prevent this outcome has yet to be determined.
For reasons outlined above, patients with ARVD should avoid strenuous physical exercise and should therefore not usually participate in competitive sports (class Ic recommendation).36,40,41
Antiarrhythmic medicationAlthough there have been few specific studies on the use of beta-blockers in ARVD, their benefits in protecting against sudden cardiac death in other conditions are well known, and their use is a class Ic recommendation. In view of the effects of exercise on the myocardium in affected individuals, the mechanism of action of beta-blockers in ARVD is likely to be by inhibiting the harmful effects of the sympathetic nervous system in this disease.
One study demonstrated that amiodarone was more effective in preventing clinically significant ventricular arrhythmias and that sotalol was associated with increased risk of first clinically relevant ventricular arrhythmia.42 However, another study showed sotalol to be superior to amiodarone and that verapamil and beta-blockers could be effective.43 Further studies are needed to determine the efficacy of these and other drugs for primary prevention in carriers of ARVD-related mutations.
In view of the conflicting data in the literature, in the case presented we decided on a low-dose beta-blocker as an adjuvant to the ICD.36,42,43
Radiofrequency ablationDue to the regional and progressive character of ARVD, radiofrequency ablation is not a definitive treatment, and should not be used in isolation or as first-line treatment. Cases in which the arrhythmogenic focus is clearly localized may benefit from the technique.44
Implantable cardioverter-defibrillatorCurrent guidelines recommend an ICD for patients fulfilling the 2010 Task Force criteria, particularly those with a family history of sudden cardiac death, sustained VT or recent unexplained syncope. The number of ventricular extrasystoles, frequency of arrhythmic events, and inducibility of VT are indicators of suitability for ICD implantation.9,12
An ICD should be considered for primary prevention in high-risk patients, although there is no agreement concerning indications. Syncope predicts appropriate ICD therapies in such patients, particularly for prevention of potentially fatal events.45,46
There is unanimity concerning the use of ICDs in the presence of documented ventricular tachycardia or fibrillation.45,47,48
The main complications of ICDs are pocket hematoma, problems with lead placement, and pericardial effusion or infection. In patients with ARVD, the RV wall may be perforated, while structural changes may hamper lead placement, reduce the device's sensitivity or affect cardiac rhythm. In addition, in young recipients the ICD will eventually need to be replaced and the leads repositioned.49
In the case presented, given the patient's unexplained syncope and induction of monomorphic VT and subsequent hemodynamic collapse, it was decided to place an ICD in line with the class Ic recommendation.47
Monitoring of relativesGenetic studies should be performed in asymptomatic relatives of AVRD patients to stratify risk, but the presence of a mutation merely indicates risk and does not necessarily mean that it will lead to clinical expression. The only formal indication for genetic testing of relatives is when a causative mutation is identified in an index case.
Studies have shown in cases of familial disease that 50% of relatives who initially show no signs of disease eventually develop it later in life.2,40,50
Diagnosis of ARVD in relatives of an affected patient is based on the presence of inverted T waves in leads V1-V3 in individuals >14 years of age (this finding may be observed in younger healthy children), late potentials on signal-averaged ECG, VT of LBBB morphology on Holter ECG monitoring or during exercise testing, >200 ventricular extrasystoles per 24 hours by Holter, and mild RV dilatation or reduced EF and regional hypokinesia.51
ConclusionAlthough there is an increasing tendency to use the term ‘arrhythmogenic cardiomyopathy’ instead of AVRD,52 due to growing awareness that LV involvement may be the dominant characteristic, this is not yet part of the 2010 Task Force diagnostic criteria. Accordingly, and since in the patient presented the left ventricle was preserved, we decided to use the traditional name for this entity throughout this report.
The present case is interesting since it illustrates several aspects mentioned in the literature. These include the clinically silent character of ARVD that means it may present late, and the marked structural deformation and RV dysfunction, reflecting a histopathologically advanced stage of the disease, in a previously asymptomatic patient.
Symptoms such as palpitations and syncope may be the manifestation of ventricular arrhythmias, which can range from frequent ventricular extrasystoles to sustained VT, their frequency being proportional to the severity of the disease.
The distribution of the disease, apparently limited to certain subpopulations, may be due to genetic factors, but is more likely to be the result of difficulties in diagnosing the condition, which probably means its true prevalence is underestimated.19
In the patient presented, who had no relevant family history, diagnosis was further hampered by confounding factors, including suspicion of acute coronary syndrome or pulmonary embolism, both of which were clinically plausible and epidemiologically more likely, and which led him to undergo various diagnostic exams that caused considerable emotional and financial hardship.
The importance of the initial ECG and echocardiography were clear, but so were the limitations of the latter in detecting RV wall motion abnormalities. MRI proved its superiority to echocardiography, and in this case was the decisive exam for the diagnosis of ARVD, even though fatty infiltration and RV wall thinning were not observed in our patient.
EPS was also important because it documented polymorphic VT, leading directly to the decision to place an ICD, even though Holter monitoring had not detected any ventricular arrhythmias.
A relationship has been identified between physical exercise and worsening of ARVD,52 which supports the idea that mechanical stress plays a part in the disease, explains the earlier and more severe presentation in athletes, and confirms the indication to restrict exercise in affected patients.
In the era of human genome sequencing, a large number of asymptomatic carriers of AVRD-related mutations who may have various stages of structural alterations are likely to be identified. For families with no identified mutation, the difficulty in arriving at the diagnosis should be cause for concern, given that sudden death can be the first manifestation of the disease.
The frequency of malignant arrhythmias as the first sign of AVRD highlights the importance of clinical awareness of the signs and symptoms that suggest the diagnosis and of early electrocardiographic and imaging screening.
Unfortunately, no reliable risk algorithm yet exists that can predict the likelihood of a particular individual developing a fatal arrhythmia and thus provide a definite indication for a prophylactic ICD. Thus, although there is no consensus on indications for an ICD, this is still the only treatment that reduces the risk of sudden death in ARVD. While some studies suggest that treatment with antiarrhythmic drugs alone is efficacious in patients with hemodynamically stable arrhythmias, current guidelines recommend an ICD for secondary prevention in patients with ventricular tachycardia or fibrillation and for primary prevention in high-risk patients (the young, athletes, and those with a strongly indicative family history or frequent syncope).45,48
In the future, the aim will be to slow progression of the disease or even to reverse it.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Marçalo J, Menezes Falcão L. Miocardiopatia arritmogénica do ventrículo direito – particularidades de um caso 2016. Rev Port Cardiol. 2017;36:217.e1–217.e10.
