
Acute aortic dissection is the most common acute aortic syndrome. It is more prevalent in males and in the elderly, and has a high mortality. Hypertension is the main risk factor. Diagnosis is based on clinical features, laboratory tests and imaging exams. Treatment is usually surgical, although in some cases an endovascular approach is an alternative.
Paraganglioma is an uncommon neuroendocrine tumor. Most produce catecholamines, and so usually manifest with hypertensive crisis, palpitations, headache and sweating. This tumor is diagnosed by measurement of plasma or urinary catecholamines and by computed tomography, magnetic resonance imaging and 123I-metaiodobenzylguanidine (MIBG) scintigraphy. Surgery is the only potentially curative treatment.
This case report describes a female patient with type A aortic dissection associated with paraganglioma. This association is very uncommon and the management of both conditions presents a challenge.
A dissecção aórtica aguda é a síndrome aórtica aguda mais frequente, ocorrendo predominantemente no sexo masculino e em idosos, estando associada a uma elevada mortalidade. Existem vários fatores de risco, destacando a hipertensão arterial. O diagnóstico é feito com base na clínica, exames laboratoriais e imagiológicos. A terapêutica habitual baseia-se na abordagem cirúrgica, existindo em alguns casos a alternativa do tratamento endovascular. O paraganglioma é um tumor neuroendócrino raro. A maioria produz catecolaminas e manifesta-se frequentemente por crises hipertensivas, palpitações, cefaleias e hipersudorese. O diagnóstico deste tumor passa pelo doseamento de catecolaminas urinárias e séricas, e pela realização de TC toracoabdominopélvica, ressonância magnética ou cintigrafia com 123MIBG. O tratamento cirúrgico é o único tratamento potencialmente curativo. Neste artigo, descreve-se um caso clínico de uma doente com uma dissecção aórtica do tipo A, associada a um paraganglioma. Esta associação é extremamente rara e a abordagem de ambas as patologias constitui um desafio.
Acute aortic dissection and paraganglioma are both rare conditions. Acute aortic dissection is defined as an intimal tear with passage of blood from the aortic lumen into the media, forming a false lumen.1 Paragangliomas are tumors that originate in extra-adrenal chromaffin cells and may or may not produce catecholamines2; they are one of the secondary causes of hypertension, which is a risk factor for aortic dissection.
We present the case of a female patient with type A aortic dissection and paraganglioma, and highlight the rarity of this association.
Case reportA 49-year-old female patient, black, born in Africa but resident in Portugal for 12 years, had a history of hypertension and hyperthyroidism. On May 20, 2010, she suffered headache, self-medicated with nebivolol and bioflavonoids. She then she suffered chest pain with no relieving factors and went to the emergency department of another hospital, where she reported two episodes of vomiting and two of hemoptysis. On observation she was alert, cooperative and oriented in time and space, hypertensive (blood pressure 200/140 mmHg) and with a heart rate of 87 bpm. There were no other abnormalities on physical examination.
Laboratory tests showed hypochromic microcytic anemia, leukocytosis (leukocytes 16200 Ul), thrombocytopenia (platelets 141×109/l), elevated D-dimers (8.01 μg/ml), low-density lipoprotein (1032 U/l), aspartate aminotransferase (60 U/l) and C-reactive protein (2.4 mg/dl), and negative troponin I (0.016 ng/ml).
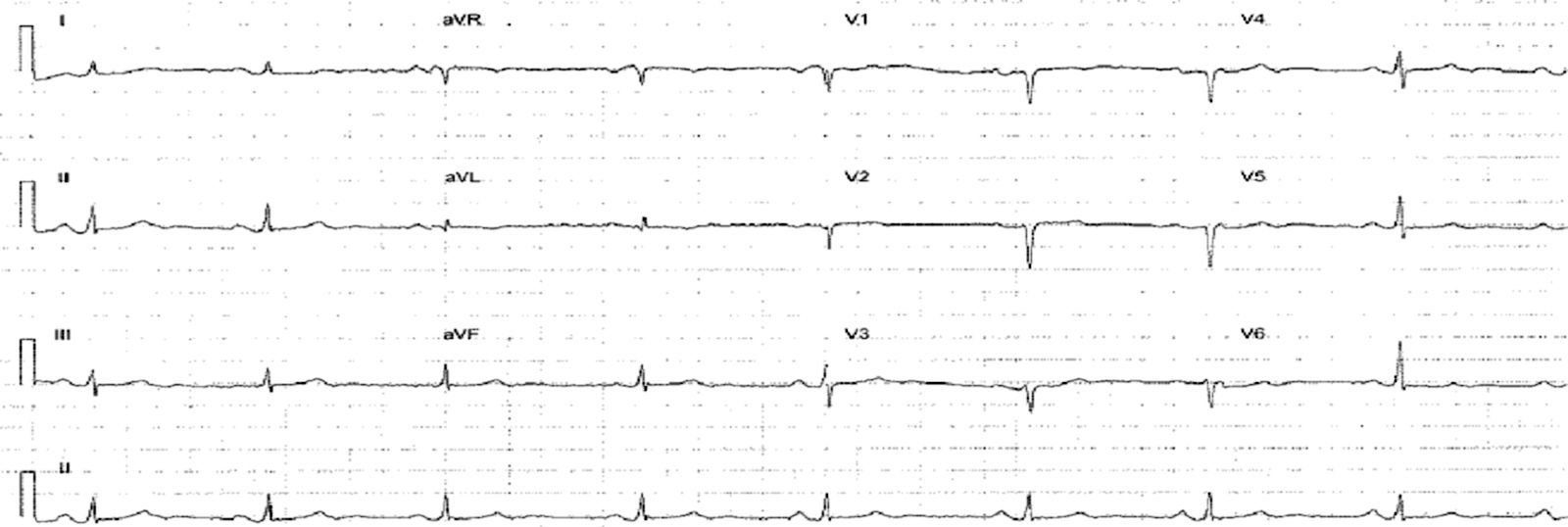
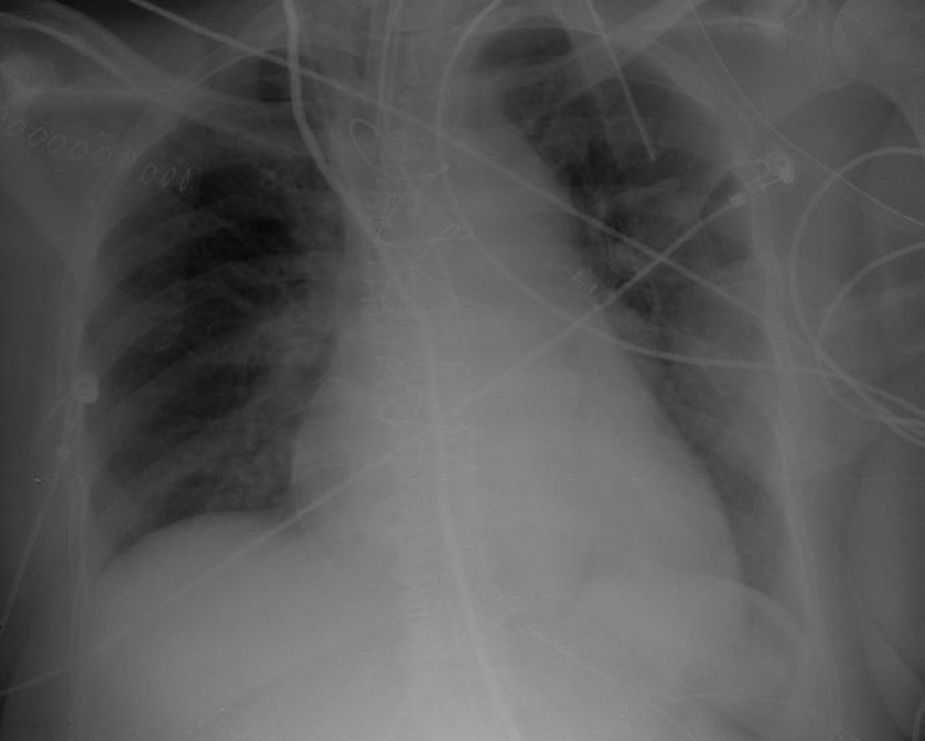
The electrocardiogram (ECG) (Figure 1) showed no significant alterations but the chest X-ray (Figure 2) revealed increased pulmonary vascularity and enlarged cardiac silhouette and mediastinum.


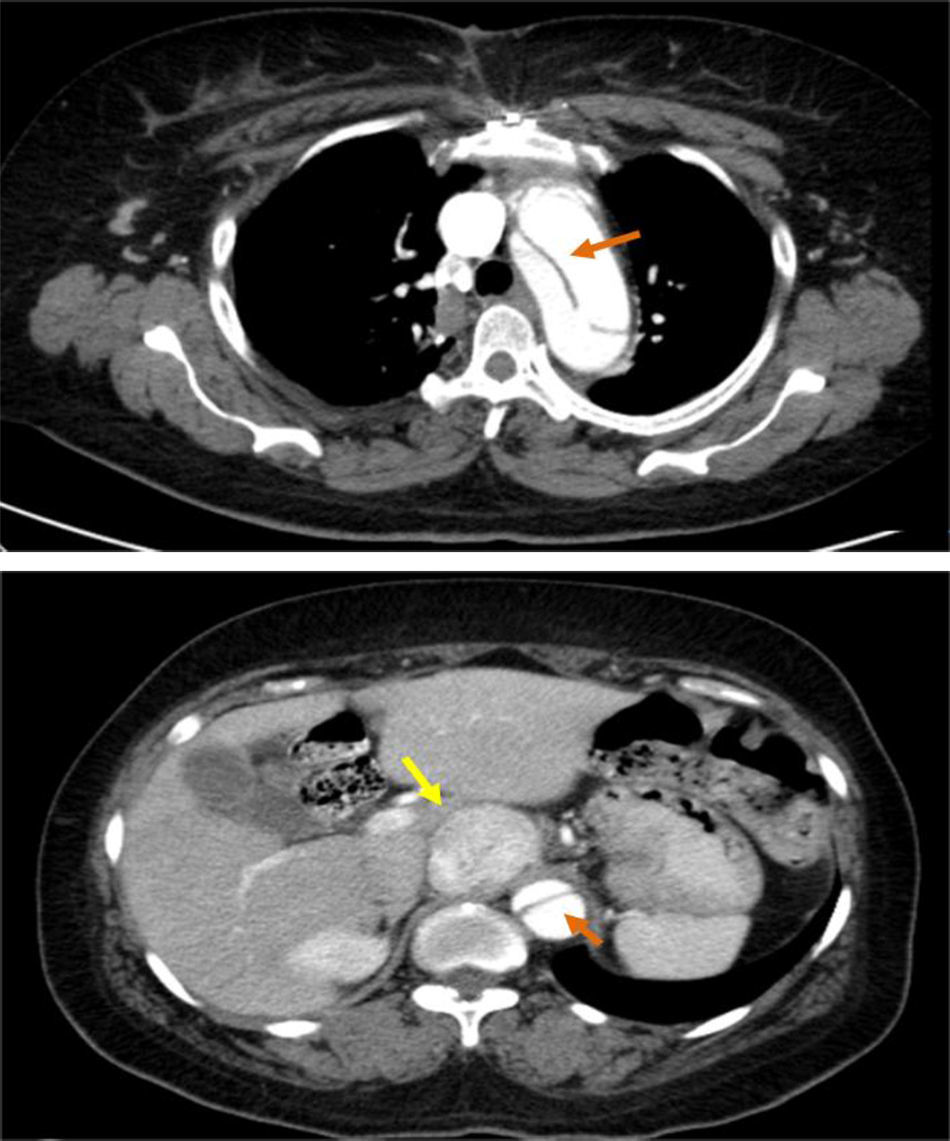
These findings prompted an urgent thoracic-abdominal-pelvic computed tomography (CT) angiogram, which showed an aneurysmal aortic dissection that extended from the aortic root to the right common iliac artery; densification of mediastinal fat with no signs of recent hemorrhage; a heterogeneous solid nodular formation around 45 mm in diameter on the right side of the aorta at the level of the emergence of the celiac trunk and in close contact with the inferior vena cava, displacing the hepatic artery anteriorly and displacing and compressing the inferior vena cava laterally; and dilatation of the proximal segment of the inferior vena cava, with a heterogeneous lumen up to the convergence of the renal veins, probably due to thrombosis (Figure 3).
 and an echodense mass compatible with a paraganglioma (yellow arrow).")
The patient was transferred to the cardiothoracic surgery department of our hospital, where she underwent urgent surgical replacement of the ascending aorta with a 26-mm Uni-Graft vascular tube graft. In the postoperative period she suffered paroxysmal hypertensive crises that prompted labetalol perfusion followed by oral alpha- and beta-blocker therapy. To clarify the clinical situation, various diagnostic exams were performed, as follows:
- •
Laboratory tests, which showed increased levels of noradrenaline (365.7 μg/24 h), normetanephrine (4268.5 μg/24 h) and evangelicalism acid (19.9 mg/24 h) in 24-h urine, and normal values of dopamine (178.1 μg/24 h), metanephrine (264.2 μg/24 h), 5-hydroxyindoleacetic acid (3.3 mg/24 h) and homovanillic acid (4.72 mg/24 h);
- •
123I-metaiodobenzylguanidine (MIBG) scintigraphy, which showed high-intensity radioisotope uptake in the median/paramedian upper abdomen, in a location corresponding to that of the nodular formation identified by CT;
- •
thoracic-abdominal-pelvic CT, which showed dissection of the aorta distal to the graft extending to the abdominal aorta up to the right common iliac artery, not involving the supra-aortic trunks, and simultaneous perfusion of the true and the false lumens; a solid, clearly circumscribed, echodense, slightly heterogeneous, nodular formation, 45 mm in its largest diameter, within the aorta and vena cava at the level of the emergence of the celiac trunk (Figure 3);
- •
Holter ECG recording and ambulatory blood pressure monitoring (ABPM), which revealed no relevant alterations;
- •
Transthoracic echocardiography, which showed concentric hypertrophy of the left ventricle and preserved global systolic function, the tube graft in the ascending aorta, and mild aortic regurgitation.
On the basis of these exams, the patient was referred to the general surgery department, where she underwent laparoscopic excision of the tumor under general anesthesia. The operation and postoperative period were uneventful and the patient's blood pressure levels remain within normal limits.
DiscussionCardiovascular disease is the leading cause of death worldwide.1 Acute aortic syndromes, which include aortic dissection, penetrating atherosclerotic ulcer and intramural hematoma, are emergent situations involving the aorta and have similar clinical characteristics and treatment.1,3–5 Acute aortic dissection is defined as an intimal tear with passage of blood from the aortic lumen into the media, forming a false lumen. It can propagate in an antegrade or retrograde fashion, and may involve the aortic branches. The presence of blood between the intima and media provokes an inflammatory response that can proceed to dilatation and rupture.1,3–5
Aortic dissection is classified as acute when <14 days after onset of symptoms, subacute (15-90 days) and chronic (>90 days).4 It is also categorized using the Stanford classification (which has replaced the DeBakey system) according to whether the ascending aorta is (type A) or is not (type B) involved.5,6
According to the Oxford Vascular Study, the incidence of acute aortic dissection is 6/100000 individuals/year, and type A dissection is more frequent than type B.7 Incidence is greater in males than in females (1.5:1), and increases with age.7 It is also more common in the winter months.5 The prognosis is poorer in women, as a result of atypical presentation and delayed diagnosis.4
Aortic dissection is associated with high morbidity and mortality. Mortality exceeds 75% within two weeks in untreated patients.3 In the Oxford Vascular Study, mortality was higher for type A dissections (73.0% at 30 days and 76.8% at five years) than for type B dissections (13.3% and 33.3%, respectively).7
The main risk factor for aortic dissection is hypertension, which is seen in 65-75% of cases, mostly uncontrolled.4,6 Other risk factors include advanced age (greater risk in the sixth and seventh decades of life); male gender; atherosclerosis; smoking (found in 61.5% of cases)7; intravenous drug abuse (cocaine and amphetamines); Valsalva maneuver; previous cardiac surgery; acute direct chest trauma; pregnancy (particularly in the third trimester) or puerperium (due to hormone-related alterations in connective tissue); congenital diseases such as Marfan, type IV Ehlers-Danlos, Loeys-Dietz, Turner, and Noonan syndromes, coarctation of the aorta, bicuspid aortic valve, or polycystic renal disease; family history of aortic disease; inflammatory disorders such as Takayasu arteritis, Behçet disease, rheumatoid arthritis and giant cell arteritis; and neuroendocrine or autoimmune disease, particularly pheochromocytoma, systemic lupus erythematosus and ankylosing spondylitis.1,3–6
Presentation is usually one of sudden intense pain, stabbing, pulsing and migratory, that may radiate to the neck or jaw, located in the anterior chest in 80% of cases, the back in 40% and the abdomen in 25%.4 The pain is frequently confused with that of acute coronary syndrome, but is not relieved by nitrates or non-opioid painkillers.6 Clinical presentation varies considerably, from hypertension or hypotension, aortic regurgitation murmur and faint pulses to syncope due to cardiac tamponade or involvement of the supra-aortic branches, or less common findings such as abdominal pain, neurological deficit, Horner syndrome, vocal cord paralysis, heart failure, pleural effusion or renal failure; 4-5% of cases are asymptomatic.1–4,8
Diagnosis is based on clinical history, physical examination and diagnostic exams.
The ECG shows non-specific alterations in most cases, while the chest X-ray is abnormal in 80% of cases, showing mediastinal enlargement.3,9
The diagnosis is confirmed by imaging studies, specifically CT, magnetic resonance imaging (MRI), echocardiography and, less frequently, aortography.1,6 Contrast CT, which has a sensitivity of 95% and specificity of 85-100%, is the most often used technique.1,4
Treatment should begin with medical therapy to control heart rate, blood pressure and symptoms and to maintain organ perfusion. The first-line agents are beta-blockers, or if these are contraindicated or not tolerated, non-dihydropyridine calcium channel blockers. If blood pressure cannot be brought under control with beta-blockers, vasodilators such as sodium nitroprusside or nitroglycerin should be used, although these should be administered only after control of heart rate. It is also important to control the pain with opiates and anxiety with benzodiazepines.1,3,6
In cases of acute type A dissection, the treatment of choice is surgery, which reduces one-month mortality from 90% to 30%.4 In uncomplicated type B dissections, the patient may be safely stabilized with medical therapy only; in complicated cases, the first-line treatment is thoracic endovascular aortic repair.3,4
Follow-up in these patients is essential, and should consist of rigorous blood pressure control and imaging studies seven days after the dissection, then at three, six and 12 months after hospital discharge. Subsequent follow-up should be yearly.1,6
Paragangliomas are neuroendocrine tumors originating in catecholamine-producing chromaffin cells of sympathetic and parasympathetic ganglia. They are distinguished from pheochromocytomas by their extra-adrenal location. Around 70% of these tumors occur in the head and neck, 20% in the abdominal and pelvic region, and 10% in the chest, where they may involve the posterior mediastinum, the para-aortic sympathetic ganglia or the heart, most often the left atrium. Paragangliomas are classified as functional or nonfunctional depending on whether they secrete catecholamines. Tumors of the parasympathetic ganglia are found almost exclusively in the neck or base of the skull in or near the carotid body and are generally nonfunctional, while tumors originating in the sympathetic ganglia may be found at any point in the sympathetic trunk, but mainly in the abdomen, and most produce catecholamines.2,10–14
Paragangliomas are rare, with an incidence of 1/300000 individuals/year,16,17 and a prevalence of 2-8 million patients/year, occurring in 0.1-0.6% of hypertensive patients. Mean age at diagnosis is 40 years.15,17
Clinical presentation is highly variable, including hypertension (90%), headache (80%), diaphoresis (71%), palpitations (64%), pallor (42%), tachycardia and anxiety. The classic triad of palpitations, headache and diaphoresis associated with hypertension has a specificity of over 90%. Fever, nausea and flushing are less common. The clinical manifestations of nonfunctional paragangliomas are due to the mass effect and include cervical mass (29%), tinnitus (23%), hearing loss (21%) and hoarseness (15%).12 Apart from hypertension, the physical examination is usually normal.11
Paroxysmal hypertensive crises occur weekly in 75% of patients. They may be triggered by anesthesia, manipulation of the tumor during surgery or by palpation or biopsy, exertion, ingestion of certain foods, and drugs, particularly corticosteroids, histamines, metoclopramide, phenothiazine, and tricyclic antidepressants.11 In 5-30% of cases the patient is asymptomatic, and it is important to exclude hypertension by ABPM. Patients with dopamine-producing tumors may be normotensive or present orthostatic hypotension.10,11,13,17 Paragangliomas may also lead to potentially fatal complications such as heart failure, myocardial infarction, arrhythmias or stroke.15
Diagnosis of paraganglioma is initially by measurement of plasma or urinary catecholamines, urinary evangelicalism acid and serum and urinary metanephrine, the O-methylated product of epinephrine. Serum metanephrine has higher sensitivity than the other tests (97% vs. 74%) but similar specificity.2,13,18
When suspicion is aroused by biochemical tests, imaging studies should be performed to confirm the diagnosis and to localize the tumor. Thoracic-abdominal-pelvic CT, with or without contrast, is the first-line exam for tumor localization, and has a sensitivity of 88-100%. MRI is indicated for patients with metastatic disease or those allergic to contrast, to avoid radiation exposure and to detect head and neck paraganglioma. Echocardiography is not recommended, since its sensitivity is low.2,13
The specificity of diagnostic exams can be improved by the use of functional techniques, particularly 123I-metaiodobenzylguanidine (MIBG) scintigraphy, which has 56-75% sensitivity and 84-100% specificity for paraganglioma.2 It is especially useful in extra-adrenal tumors, those larger than 5 cm, multifocal disease and malignancy.13 In patients with metastatic disease, 18-fluorodeoxyglucose positron emission tomography (PET) is preferable to MIBG scintigraphy, although it should not be used for initial diagnosis.13 Other compounds used in PET scanning include 6-[18F]fluorodopamine, 6-[18F]fluorodopa, and 11C-hydroxyephedrine.13
Hypertension should be controlled with both alpha-adrenergic and beta-adrenergic blockers. Beta-blockers should not be used in isolation, due to the risk of triggering a hypertensive crisis by increasing vasoconstriction. Calcium channel blockers are used as second-line therapy. Magnesium sulfate can be administered in cases of hypertension resistant to conventional treatment.11
Surgery is the only potentially curative treatment.2,13,15,19 However, surgery triggers the release of catecholamines, causing complications that can be fatal.13 In order to ensure that surgery is safe, appropriate preoperative preparation is required, consisting of alpha-blocker administration for 10-14 days and, when adequate alpha blockade is achieved, beta-blockers for 2-5 days before the operation. An alternative regimen is calcium channel blockers or the dual alpha- and beta-adrenergic blocker labetalol. Treatment should also include a sodium-rich diet and abundant oral fluid intake to avoid severe hypotension after the tumor is removed.2,11,13
Postoperatively, the main complications are hypotension and hypoglycemia. Antihypertensive medication should be reduced gradually to stabilize blood pressure.2,15
In incurable cases, with very large tumors and metastatic disease, the therapeutic options are chemotherapy and radiotherapy. Molecular therapy with sunitinib, a tyrosine kinase inhibitor, is an upcoming therapeutic option.19
To verify complete removal of the tumor, biochemical tests should be repeated 2-4 weeks after surgery.2 Patients should be followed indefinitely, with monitoring of blood pressure and urinary and serum catecholamine levels.2,13,15
One of the most serious complications of pheochromocytoma and paraganglioma is aortic dissection.20,21 The catecholamines produced by these tumors cause rises in blood pressure and hence wall stress in the aorta, which can lead to acute dissection.
The case reported here is interesting for the rarity of the association of the two conditions, of which there are few descriptions in the literature. The first case of a patient with dissecting aortic aneurysm and pheochromocytoma was reported in 1975 by Triplett and Atuk,22 and Azizi et al. reported a case of concomitant type A dissection and pheochromocytoma.22 Cases have been reported of pheochromocytoma associated with dissecting aneurysm of the abdominal aorta and with carotid artery dissection.21
One of the main problems in this association is how to treat the two conditions. Aortic dissection is a surgical emergency, with high mortality if untreated, but the concomitant presence of pheochromocytoma or paraganglioma complicates anesthetic induction and increases surgical risk. A multidisciplinary approach is thus essential.22
In the case presented, the dissection was rapidly and successfully corrected without effects related to the paraganglioma, and then following alpha and beta blockade the tumor was removed without complications.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Borrego A, Carrilho Ferreira P, Pinto FJ. Dissecção aórtica aguda do tipo A em doente com paraganglioma. Rev Port Cardiol. 2017;36:777.e1–777.e6.
